| Protein: | RNH70 |
| Organism: | Saccharomyces cerevisiae |
| Length: | 553 amino acids |
| Reference: | Malmström L, et al. (2007) Superfamily assignments for the yeast proteome through integration of structure prediction with the gene ontology. PLoS Biol 5(4): e76. doi:10.1371/journal.pbio.0050076 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for RNH70.
| Description | E-value | Query Range |
Subject Range |
|
|
436.0 | [0..23] | [553..41] |
|
|
426.0 | [0..18] | [532..4] |
|
|
319.0 | [0..38] | [502..141] |
|
|
288.0 | [0..45] | [460..38] |
|
|
285.0 | [0..45] | [460..38] |
|
|
281.0 | [0..45] | [460..38] |
|
|
281.0 | [0..60] | [553..151] |
|
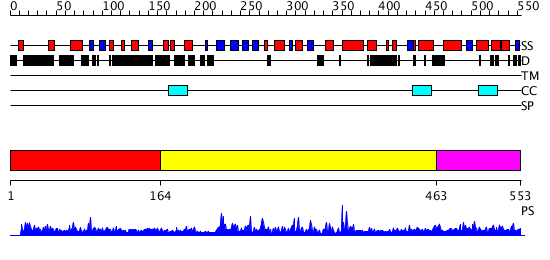
Region A: Residues: [1-163] |
1 11 21 31 41 51
| | | | | |
1 MQVEGPDTNF VSDLALGSKK RRLSKTSVQE DDHTNVVSEV NKNKKKKKAK PMTCTLLKSV 60
61 VEKGIGIKDV RDMTQYLLQA ENNSPKWIDI CNRSSLQKMI VLFIPGLQPD DFENGKNTFN 120
121 EISDDNFKYI PGEIASTFHT FPVMAPGSKM TLFSPYNSFI NVG
|
| Detection Method: |
Shown below are all of our structure predictions for this domain.
Click here to view only most confident match.
Found no structure predictions for this domain.
|
Region A: Residues: [164-462] |
1 11 21 31 41 51
| | | | | |
1 LSKMEKINKL KELQKKKKIT INDLVLSEQQ LVANDYPLDS GDTNFDTDWV QTVDFTHGGS 60
61 HIFALDCEMC LSEQGLVLTR ISLVNFDNEV IYEELVKPDV PIVDYLTRYS GITEEKLTVG 120
121 AKKTLREVQK DLLKIISRSD ILIGHSLQND LKVMKLKHPL VVDTAIIYHH KAGDPFKPSL 180
181 KYLSETFLNK SIQNGEHDSV EDARACLELT KLKILNGLAF GIGINTENLF TKLHRFEVKT 240
241 VLLNDMIIKN HTEDDSKGQL IRCVEDDETW THIHENLNKD VKLIVGRIKN LERSRNYNK
|
| Detection Method: | |
| Confidence: | 10.30103 |
| Match: | 1dpiA |
| Description: | STRUCTURE OF LARGE FRAGMENT OF ESCHERICHIA COLI DNA POLYMERASE I COMPLEXED WITH D/TMP |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [463-553] |
1 11 21 31 41 51
| | | | | |
1 KPRKETPSFD ASMVLHDIGQ HLTQLYENAT PGTMILIMSG TGDTRPWNNL STELEFIQDK 60
61 KERLDKRRER EPEIVEAIKL ARGGVASFTV K
|
| Detection Method: |
Shown below are all of our de novo (Rosetta) predictions for this domain.
Click here to view only most confident match.
| MCM Score |
SCOP Match |
SCOP Description | ||
| View | Download | 0.539 | a.116.1 | GTPase activation domain, GAP |
| View | Download | 0.539 | a.116.1 | GTPase activation domain, GAP |
| View | Download | 0.506 | c.58.1 | Aminoacid dehydrogenase-like, N-terminal domain |
| View | Download | 0.506 | c.58.1 | Aminoacid dehydrogenase-like, N-terminal domain |
| View | Download | 0.499 | c.47.1 | Thioredoxin-like |
| View | Download | 0.447 | a.47.2 | t-snare proteins |
| View | Download | 0.431 | c.44.2 | Enzyme IIB-cellobiose |
| View | Download | 0.431 | c.44.2 | Enzyme IIB-cellobiose |
| View | Download | 0.359 | a.29.4 | RecG, N-terminal domain |
| View | Download | 0.355 | a.7.1 | Spectrin repeat |