| Protein: | MGA1 |
| Organism: | Saccharomyces cerevisiae |
| Length: | 456 amino acids |
| Reference: | Malmström L, et al. (2007) Superfamily assignments for the yeast proteome through integration of structure prediction with the gene ontology. PLoS Biol 5(4): e76. doi:10.1371/journal.pbio.0050076 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for MGA1.
| Description | E-value | Query Range |
Subject Range |
|
|
0.0 | [4..439] | [8..457] |
|
|
0.0 | [4..439] | [8..453] |
|
|
0.0 | [4..433] | [16..485] |
|
|
0.0 | [4..433] | [16..485] |
|
|
0.0 | [5..432] | [22..448] |
|
|
0.0 | [4..428] | [19..458] |
|
|
0.0 | [4..428] | [19..458] |
|
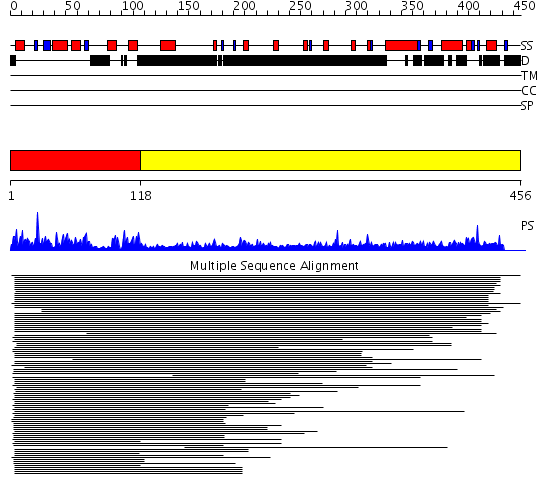
Region A: Residues: [1-117] |
1 11 21 31 41 51
| | | | | |
1 MQPKTFVHQL HAILLEPEVN KWIYWSPTDN TVFFLKPYDP NFSTHVLKRY FKHGNVNSFV 60
61 RQLHMYGFHK LSHPSPDQSS ANNGNVKELV EWKFTHPSGF FFKEANAGIL NKIQRKS
|
| Detection Method: | |
| Confidence: | 129.70927 |
| Match: | 1hks__ |
| Description: | Heat-shock transcription factor |
Matching Structure (courtesy of the PDB): |
|
| Term | Confidence | Notes |
| transcription regulator activity | 2.69048535857291 | bayes_pls_golite062009 |
| nucleic acid binding | 2.64324711228084 | bayes_pls_golite062009 |
| DNA binding | 2.61442118042246 | bayes_pls_golite062009 |
| binding | 2.37880408751912 | bayes_pls_golite062009 |
| transcription factor activity | 1.88900792328916 | bayes_pls_golite062009 |
| sequence-specific DNA binding | 0.68922344928172 | bayes_pls_golite062009 |
| transcription activator activity | 0.485327753327262 | bayes_pls_golite062009 |
| transcription repressor activity | 0.385147412723772 | bayes_pls_golite062009 |
| protein binding | 0.35301104350965 | bayes_pls_golite062009 |
|
Region A: Residues: [118-456] |
1 11 21 31 41 51
| | | | | |
1 TGVGKDGKRK NILSPISVSY VDASRLNVLS QQSGPVSARE PSNMFMGSPV HYSTSQSPPH 60
61 ISIPQQQQSS GPYLISSLPP QQPTVNMMRR QSISARMMNS YDYPNQFSTQ DSIVQPQQPQ 120
121 QVLSPQALSG PPMKKSGTLS STDDLKTTSL PIVNYPMPYH PGAFAQQQQQ QQQPLPTVPP 180
181 YSSYSTPFPS MMNSLSNSAS NSPALGVCNN NVTLPKKSNI SERQALDNHI QTLKNSLSTI 240
241 TDLIEKHINS ASQDENKTLT NDAMNKDLRT SLSLLQNSKE EIIQLESKWM SMQSVKTTAL 300
301 PLQETTNTSS TLTSLTSSII PKSIPIITKG EVATKPASY
|
| Detection Method: | |
| Confidence: | 14.93 |
| Match: | 1i3qA |
| Description: | RBP1 |
Matching Structure (courtesy of the PDB): |
|