| Protein: | PWP2_YEAST |
| Organism: | Saccharomyces cerevisiae S288c |
| Length: | 923 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for PWP2_YEAST.
| Description | E-value | Query Range |
Subject Range |
|
|
0.0 | [1..923] | [1..923] |
|
|
0.0 | [15..740] | [865..1515] |
|
|
0.0 | [1..896] | [1..897] |
|
|
0.0 | [1..853] | [1..846] |
|
|
0.0 | [1..862] | [1..888] |
|
|
0.0 | [1..897] | [1..934] |
|
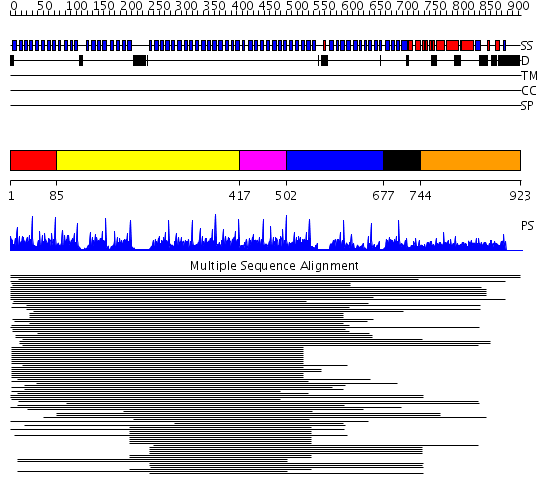
Region A: Residues: [1-84] |
1 11 21 31 41 51
| | | | | |
1 MKSDFKFSNL LGTVYRQGNI TFSDDGKQLL SPVGNRVSVF DLINNKSFTF EYEHRKNIAA 60
61 IDLNKQGTLL ISIDEDGRAI LVNF
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [85-416] |
1 11 21 31 41 51
| | | | | |
1 KARNVLHHFN FKEKCSAVKF SPDGRLFALA SGRFLQIWKT PDVNKDRQFA PFVRHRVHAG 60
61 HFQDITSLTW SQDSRFILTT SKDLSAKIWS VDSEEKNLAA TTFNGHRDYV MGAFFSHDQE 120
121 KIYTVSKDGA VFVWEFTKRP SDDDDNESED DDKQEEVDIS KYSWRITKKH FFYANQAKVK 180
181 CVTFHPATRL LAVGFTSGEF RLYDLPDFTL IQQLSMGQNP VNTVSVNQTG EWLAFGSSKL 240
241 GQLLVYEWQS ESYILKQQGH FDSTNSLAYS PDGSRVVTAS EDGKIKVWDI TSGFCLATFE 300
301 EHTSSVTAVQ FAKRGQVMFS SSLDGTVRAW DL
|
| Detection Method: | |
| Confidence: | 131.274545 |
| Match: | 1gxrA_ |
| Description: | Groucho/tle1, C-terminal domain |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [417-501] |
1 11 21 31 41 51
| | | | | |
1 IRYRNFRTFT GTERIQFNCL AVDPSGEVVC AGSLDNFDIH VWSVQTGQLL DALSGHEGPV 60
61 SCLSFSQENS VLASASWDKT IRIWS
|
| Detection Method: | |
| Confidence: | 252.69897 |
| Match: | 1gg2B_ |
| Description: | beta1-subunit of the signal-transducing G protein heterotrimer |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [502-676] |
1 11 21 31 41 51
| | | | | |
1 IFGRSQQVEP IEVYSDVLAL SMRPDGKEVA VSTLKGQISI FNIEDAKQVG NIDCRKDIIS 60
61 GRFNQDRFTA KNSERSKFFT TIHYSFDGMA IVAGGNNNSI CLYDVPNEVL LKRFIVSRNM 120
121 ALNGTLEFLN SKKMTEAGSL DLIDDAGENS DLEDRIDNSL PGSQRGGDLS TRKMR
|
| Detection Method: | |
| Confidence: | 202.9897 |
| Match: | 1erjA_ |
| Description: | Tup1, C-terminal domain |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [677-743] |
1 11 21 31 41 51
| | | | | |
1 PEVRVTSVQF SPTANAFAAA STEGLLIYST NDTILFDPFD LDVDVTPHST VEALREKQFL 60
61 NALVMAF
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [744-923] |
1 11 21 31 41 51
| | | | | |
1 RLNEEYLINK VYEAIPIKEI PLVASNIPAI YLPRILKFIG DFAIESQHIE FNLIWIKALL 60
61 SASGGYINEH KYLFSTAMRS IQRFIVRVAK EVVNTTTDNK YTYRFLVSTD GSMEDGAADD 120
121 DEVLLKDDAD EDNEENEEND VVMESDDEEG WIGFNGKDNK LPLSNENDSS DEEENEKELP 180
181
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.