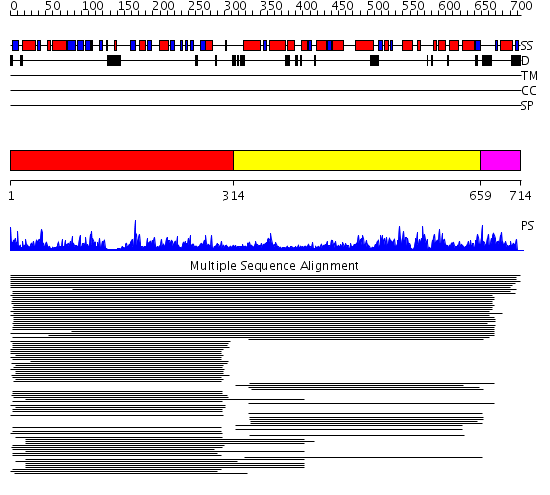
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for gi|207344678.
| Description |
E-value |
Query
Range |
Subject
Range |
NADSYN1 - NAD synthetase 1
|
0.0 |
[1..709] |
[1..703] |
gi|207344678 - gi|207344678|gb|EDZ71741.1| YHR074Wp-like protein [Saccharomyces cerevisiae AWRI1631]
NADE_YEAST - Glutamine-dependent NAD(+) synthetase OS=Saccharomyces cerevisiae (strain ATCC 204508 / S288c) GN=QN...
gi|151944019 - gi|151944019|gb|EDN62312.1| glutamine-dependent NAD synthetase [Saccharomyces cerevisiae YJM789]
QNS1 - Glutamine-dependent NAD(+) synthetase, essential for the formation of NAD(+) from nicotinic acid ade...
|
0.0 |
[1..714] |
[1..714] |
NADE_MOUSE - Glutamine-dependent NAD(+) synthetase OS=Mus musculus GN=Nadsyn1 PE=1 SV=1
gi|40644110 - gi|40644110|emb|CAC83797.1| NH3-dependent NAD+ synthetase like protein [Mus musculus domesticus]
|
0.0 |
[1..707] |
[1..701] |
NADE_SCHPO - Putative glutamine-dependent NAD(+) synthetase OS=Schizosaccharomyces pombe (strain 972 / ATCC 24843...
SPCC553.02 - glutamine-dependent NAD(+) synthetase
|
0.0 |
[1..704] |
[1..698] |
CG9940-PA, CG994... - This gene is referred to in FlyBase by the symbol Dmel\CG9940 (CG9940, FBgn0030512). It is a protein...
|
0.0 |
[1..701] |
[1..696] |
NADE_ARATH - Glutamine-dependent NAD(+) synthetase OS=Arabidopsis thaliana GN=At1g55090 PE=2 SV=1
|
0.0 |
[3..700] |
[2..693] |
CE18521 - status:Confirmed UniProt:Q9XXK6 protein_id:CAA18773.1
|
0.0 |
[3..707] |
[6..703] |
NAE2_THEMA, NADE... - (Q9X0Y0) Probable glutamine-dependent NAD(+) synthetase (EC 6.3.5.1) (NAD(+) synthase [glutamine-hyd...
NADE2_THEMA - Probable glutamine-dependent NAD(+) synthetase OS=Thermotoga maritima (strain ATCC 43589 / MSB8 / DS...
|
0.0 |
[1..675] |
[1..572] |
gi|25500547 - pir||G97029 nH(3)-dependent NAD(+) synthetase [imported] - Clostridium acetobutylicum
gi|15894337, gi|... - gi|15894337|ref|NP_347686.1| NAD synthetase [Clostridium acetobutylicum ATCC 824], gi|15023963|gb|AA...
|
0.0 |
[3..678] |
[4..625] |
NADE_AQUAE - Probable glutamine-dependent NAD(+) synthetase OS=Aquifex aeolicus GN=nadE PE=3 SV=1
NADE_AQUAE - Probable glutamine-dependent NAD(+) synthetase OS=Aquifex aeolicus (strain VF5) GN=nadE PE=3 SV=1
|
0.0 |
[4..678] |
[1..558] |