| Protein: | HSFB1_ARATH |
| Organism: | Arabidopsis thaliana |
| Length: | 284 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for HSFB1_ARATH.
| Description | E-value | Query Range |
Subject Range |
|
|
309.0 | [0..1] | [284..1] |
|
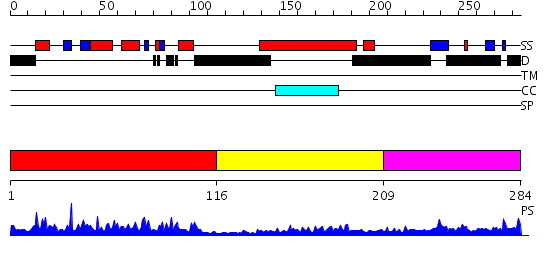
Region A: Residues: [1-115] |
1 11 21 31 41 51
| | | | | |
1 MTAVTAAQRS VPAPFLSKTY QLVDDHSTDD VVSWNEEGTA FVVWKTAEFA KDLLPQYFKH 60
61 NNFSSFIRQL NTYGFRKTVP DKWEFANDYF RRGGEDLLTD IRRRKSVIAS TAGKC
|
| Detection Method: | |
| Confidence: | 33.221849 |
| Match: | 1hksA |
| Description: | SOLUTION STRUCTURE OF THE DNA-BINDING DOMAIN OF DROSOPHILA HEAT SHOCK TRANSCRIPTION FACTOR |
Matching Structure (courtesy of the PDB): |
|
| Term | Confidence | Notes |
| binding | 0.835523092977445 | bayes_pls_golite062009 |
| transcription regulator activity | 0.4843036104266 | bayes_pls_golite062009 |
| nucleic acid binding | 0.37117756729668 | bayes_pls_golite062009 |
| DNA binding | 0.3118855114645 | bayes_pls_golite062009 |
|
Region A: Residues: [116-208] |
1 11 21 31 41 51
| | | | | |
1 VVVGSPSESN SGGGDDHGSS STSSPGSSKN PGSVENMVAD LSGENEKLKR ENNNLSSELA 60
61 AAKKQRDELV TFLTGHLKVR PEQIDKMIKG GKF
|
| Detection Method: | |
| Confidence: | 1.0 |
| Match: | 1hjbA |
| Description: | C/ebp beta |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [209-284] |
1 11 21 31 41 51
| | | | | |
1 KPVESDEESE CEGCDGGGGA EEGVGEGLKL FGVWLKGERK KRDRDEKNYV VSGSRMTEIK 60
61 NVDFHAPLWK SSKVCN
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.