| Protein: | rsc1 |
| Organism: | Schizosaccharomyces pombe |
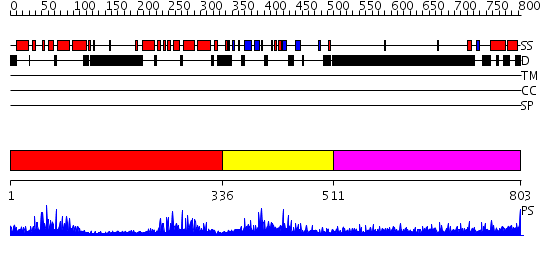
| Length: | 803 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for rsc1.
| Description | E-value | Query Range |
Subject Range |
|
|
527.0 | [0..30] | [799..115] |
|
|
473.0 | [0..16] | [777..37] |
|
|
469.0 | [0..14] | [796..118] |
|
|
466.0 | [0..8] | [684..3] |
|
|
449.0 | [0..14] | [729..180] |
|
|
444.0 | [0..14] | [682..3] |
|
|
433.0 | [0..16] | [774..38] |
|
|
433.0 | [0..16] | [790..38] |
|
|
429.0 | [0..14] | [717..139] |
|
Region A: Residues: [1-335] |
1 11 21 31 41 51
| | | | | |
1 MSSKIRPSAD DKKLQRVLYF FLERVRAAKD VSGQLLSPLI DNASVDTASV SPSSNGRPTT 60
61 LKSIQSKIDE FQYHDFSEFV SDLAYLFINV KALYEGTQTY SFVQALEEFC IQQLRTFQQQ 120
121 GYIPVITWPN TDSPSATTSS PISRNPEYSV SPPNGSKFVK NEDEAYDSDL YVEEEDSDVK 180
181 GRSMVGRDGR YKSEDLKRRK LQPSSKPLSS LEARAKVIMR QVRRYRDGSG RQLFAPFERL 240
241 PDPRMFPEYY QAIEQPMALE VIQKKLSKHR YETIEQFVDD FNLMFDNAKS FNDPSSQVYR 300
301 DADFLKNYLA DVLRLEAGKL DSEFFNYETD SRASP
|
| Detection Method: | |
| Confidence: | 30.154902 |
| Match: | 1eqfA |
| Description: | TAFII250 double bromodomain module |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [336-510] |
1 11 21 31 41 51
| | | | | |
1 QLPKNDIQPA VSIDGTLLNV GDWVLIRNPA DSSKPIVSQI YRIWKSDDDI NYVTVCWYLR 60
61 PEQTVHRADA VFYENEVFKT SLYRDHPVSE IVGRCFVMYI TRYIRGRPKG IRSTPVFVCE 120
121 SRYNDDTKQF SKIKSWKACM PQEVSGSEYE MILFDRPITL TKVASPLLHL LASKS
|
| Detection Method: | |
| Confidence: | 49.522879 |
| Match: | 1w4sA |
| Description: | Crystal structure of the proximal BAH domain of polybromo |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [511-803] |
1 11 21 31 41 51
| | | | | |
1 QGLPSPATTD SNTHMLPSQG SLLPPSSISE TKSFSTKAST PLSTDDIATP LSSAPNPPSV 60
61 MPTYARKTSS HSERSSHSSY HNSSHVPTAA FNSPIMRTST KSTSPIPARP FYAQSGSLQS 120
121 LNTTQHSHQI SGGHSGRMNV PYAKLSYTSH NGRHGGSNGN ISGAKTPMTN YTINSMPSLP 180
181 VFPPAFIVPG THQKLDESSP VPGIDDVTVI NTETAKMLDK DEHQNVLWYT VPPLDPIPLE 240
241 NRNGSLTHSV EYVLYKKSKG SQVITEKARS NELSREAKFE NLVASLSDAL IPP
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.