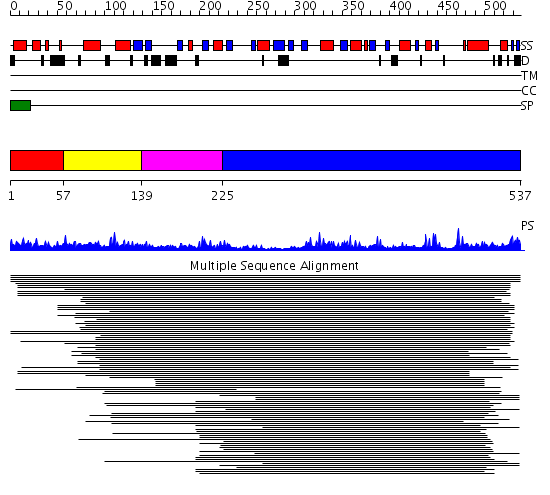
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for APE3.
| Description |
E-value |
Query
Range |
Subject
Range |
gi|7507776 - gi|7507776|pir||T16867 probable cytochrome P450 T13C5.1 [similarity] - Caenorhabditis elegans
|
0.0 |
[1..537] |
[28..564] |
APE3 - Vacuolar aminopeptidase Y, processed to mature form by Prb1p
APE3_YEAST - Aminopeptidase Y OS=Saccharomyces cerevisiae (strain ATCC 204508 / S288c) GN=APE3 PE=1 SV=1
|
0.0 |
[1..537] |
[1..537] |
TFR1_MOUSE - Transferrin receptor protein 1 OS=Mus musculus GN=Tfrc PE=1 SV=1
|
3.0E-77 |
[6..527] |
[69..631] |
TFR1_CANLF - Transferrin receptor protein 1 OS=Canis lupus familiaris GN=TFRC PE=1 SV=1
|
4.0E-72 |
[8..527] |
[74..638] |
gi|25486574 - pir||E84132 aminopeptidase BH3861 [imported] - Bacillus halodurans (strain C-125)
gi|10176486, gi|... - gi|15616423|ref|NP_244728.1| aminopeptidase [Bacillus halodurans C-125], gi|10176486|dbj|BAB07580.1|...
|
7.0E-71 |
[57..526] |
[21..458] |
TFR1_FELCA - Transferrin receptor protein 1 OS=Felis catus GN=TFRC PE=1 SV=1
|
9.0E-71 |
[8..523] |
[74..633] |
TFRC - transferrin receptor (p90, CD71)
|
1.0E-69 |
[8..527] |
[71..628] |
gi|16033408 - gi|16033408|gb|AAL13231.1|AF358651_1 leucine aminopeptidase precursor [Bacillus licheniformis]
|
2.0E-63 |
[76..510] |
[33..444] |
gi|121491979, gi... - gi|121636332|ref|YP_976555.1| putative lipoprotein aminopeptidase lpqL [Mycobacterium bovis BCG str....
gi|148821614, gi... - gi|148821614|ref|YP_001286368.1| lipoprotein aminopeptidase lpqL [Mycobacterium tuberculosis F11], g...
gi|255098386 - gi|255098386|ref|ZP_05327864.1| putative lipoprotein aminopeptidase [Mycobacterium tuberculosis KZN ...
gi|254817417 - gi|254817417|ref|ZP_05222418.1| putative lipoprotein aminopeptidase [Mycobacterium tuberculosis KZN ...
gi|254549364 - gi|254549364|ref|ZP_05139811.1| lipoprotein aminopeptidase lpqL [Mycobacterium tuberculosis '98-R604...
gi|253318845, gi... - gi|253797342|ref|YP_003030343.1| lipoprotein aminopeptidase lpqL [Mycobacterium tuberculosis KZN 143...
gi|124599828, gi... - gi|81255723|ref|ZP_00880193.1| COG2234: Predicted aminopeptidases [Mycobacterium tuberculosis C], gi...
gi|31791596, gi|... - gi|31791596|ref|NP_854089.1| lipoprotein aminopeptidase LpqL [Mycobacterium bovis AF2122/97], gi|316...
LPQL_MYCTU - Probable lipoprotein aminopeptidase LpqL OS=Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) G...
gi|215409931 - gi|215409931|ref|ZP_03418739.1| lipoprotein aminopeptidase lpqL [Mycobacterium tuberculosis 94_M4241...
gi|254363382, gi... - gi|254363382|ref|ZP_04979428.1| lipoprotein aminopeptidase lpqL [Mycobacterium tuberculosis str. Haa...
gi|215425639 - gi|215425639|ref|ZP_03423558.1| lipoprotein aminopeptidase lpqL [Mycobacterium tuberculosis T92]
gi|148660183, gi... - gi|167970766|ref|ZP_02553043.1| lipoprotein aminopeptidase lpqL [Mycobacterium tuberculosis H37Ra], ...
gi|219556238 - gi|219556238|ref|ZP_03535314.1| lipoprotein aminopeptidase lpqL [Mycobacterium tuberculosis T17]
gi|224988804, gi... - gi|224988804|ref|YP_002643491.1| putative lipoprotein aminopeptidase [Mycobacterium bovis BCG str. T...
gi|7477880 - pir||H70629 probable AMINOPEPTIDASE - Mycobacterium tuberculosis (strain H37RV)�
|
2.0E-60 |
[74..518] |
[47..494] |