| Protein: | UTP21 |
| Organism: | Saccharomyces cerevisiae |
| Length: | 939 amino acids |
| Reference: | Malmström L, et al. (2007) Superfamily assignments for the yeast proteome through integration of structure prediction with the gene ontology. PLoS Biol 5(4): e76. doi:10.1371/journal.pbio.0050076 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for UTP21.
| Description | E-value | Query Range |
Subject Range |
|
|
755.0 | [0..2] | [788..756] |
|
|
713.0 | [0..19] | [713..859] |
|
|
713.0 | [0..13] | [657..1161] |
|
|
661.0 | [0..2] | [622..1063] |
|
|
660.0 | [0..7] | [654..937] |
|
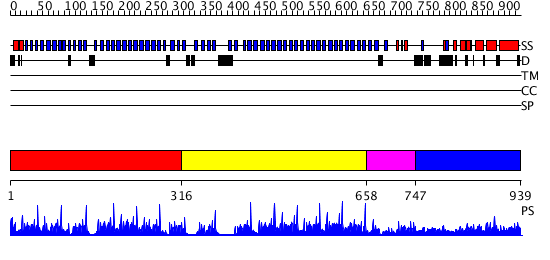
Region A: Residues: [1-315] |
1 11 21 31 41 51
| | | | | |
1 MSIDLKKRKV EEDVRSRGKN SKIFSPFRII GNVSNGVPFA TGTLGSTFYI VTCVGKTFQI 60
61 YDANTLHLLF VSEKETPSSI VALSAHFHYV YAAYENKVGI YKRGIEEHLL ELETDANVEH 120
121 LCIFGDYLCA STDDNSIFIY KKSDPQDKYP SEFYTKLTVT EIQGGEIVSL QHLATYLNKL 180
181 TVVTKSNVLL FNVRTGKLVF TSNEFPDQIT TAEPAPVLDI IALGTVTGEV IMFNMRKGKR 240
241 IRTIKIPQSR ISSLSFRTDG SSHLSVGTSS GDLIFYDLDR RSRIHVLKNI HRESYGGVTQ 300
301 ATFLNGQPII VTSGG
|
| Detection Method: | |
| Confidence: | 127.69897 |
| Match: | 1gg2B |
| Description: | beta1-subunit of the signal-transducing G protein heterotrimer |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [316-657] |
1 11 21 31 41 51
| | | | | |
1 DNSLKEYVFD PSLSQGSGDV VVQPPRYLRS RGGHSQPPSY IAFADSQSHF MLSASKDRSL 60
61 WSFSLRKDAQ SQEMSQRLHK KQDGGRVGGS TIKSKFPEIV ALAIENARIG EWENIITAHK 120
121 DEKFARTWDM RNKRVGRWTF DTTDDGFVKS VAMSQCGNFG FIGSSNGSIT IYNMQSGILR 180
181 KKYKLHKRAV TGISLDGMNR KMVSCGLDGI VGFYDFNKST LLGKLKLDAP ITAMVYHRSS 240
241 DLFALALDDL SIVVIDAVTQ RVVRQLWGHS NRITAFDFSP EGRWIVSASL DSTIRTWDLP 300
301 TGGCIDGIIV DNVATNVKFS PNGDLLATTH VTGNGICIWT NR
|
| Detection Method: | |
| Confidence: | 110.9794 |
| Match: | 1gxrA |
| Description: | Groucho/tle1, C-terminal domain |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [658-746] |
1 11 21 31 41 51
| | | | | |
1 AQFKTVSTRT IDESEFARMA LPSTSVRGND SMLSGALESN GGEDLNDIDF NTYTSLEQID 60
61 KELLTLSIGP RSKMNTLLHL DVIRKRSKP
|
| Detection Method: | |
| Confidence: | 135.54902 |
| Match: | 1erjA |
| Description: | Tup1, C-terminal domain |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [747-939] |
1 11 21 31 41 51
| | | | | |
1 KEAPKKSEKL PFFLQLSGEK VGDEASVREG IAHETPEEIH RRDQEAQKKL DAEEQMNKFK 60
61 VTGRLGFESH FTKQLREGSQ SKDYSSLLAT LINFSPAAVD LEIRSLNSFE PFDEIVWFID 120
121 ALTQGLKSNK NFELYETFMS LLFKAHGDVI HANNKNQDIA SALQNWEDVH KKEDRLDDLV 180
181 KFCMGVAAFV TTA
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
| MCM Score |
GO Score |
GO Term |
SCOP Match |
SCOP Description | ||
| View | Download | 0.820 | N/A | N/A | a.118.8 | TPR-like |