| Protein: | MMSA_ARATH |
| Organism: | Arabidopsis thaliana |
| Length: | 607 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for MMSA_ARATH.
| Description | E-value | Query Range |
Subject Range |
|
|
597.0 | [0..95] | [592..4] |
|
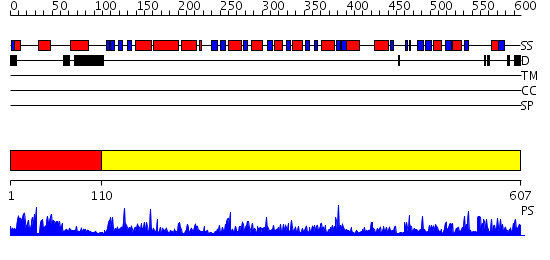
Region A: Residues: [1-109] |
1 11 21 31 41 51
| | | | | |
1 MVRVKQKNLE SYRSNGTYPP TWRNPTTSFA PDQHRVSIHS SLKSKTKRRR LYKEADDNTK 60
61 LRSSSSTTTT TTTMLLRISG NNLRPLRPQF LALRSSWLST SPEQSTQPQ
|
| Detection Method: | |
| Confidence: | 37.30103 |
| Match: | 1tiwA |
| Description: | Crystal structure of E. coli PutA proline dehydrogenase domain (residues 86-669) complexed with L-Tetrahydro-2-furoic acid |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [110-607] |
1 11 21 31 41 51
| | | | | |
1 MPPRVPNLIG GSFVESQSSS FIDVINPATQ EVVSKVPLTT NEEFKAAVSA AKQAFPLWRN 60
61 TPITTRQRVM LKFQELIRKN MDKLAMNITT EQGKTLKDSH GDIFRGLEVV EHACGMATLQ 120
121 MGEYLPNVSN GVDTYSIREP LGVCAGICPF NFPAMIPLWM FPVAVTCGNT FILKPSEKDP 180
181 GASVILAELA MEAGLPDGVL NIVHGTNDTV NAICDDEDIR AVSFVGSNTA GMHIYARAAA 240
241 KGKRIQSNMG AKNHGLVLPD ANIDATLNAL LAAGFGAAGQ RCMALSTVVF VGDAKSWEDK 300
301 LVERAKALKV TCGSEPDADL GPVISKQAKE RICRLIQSGV DDGAKLLLDG RDIVVPGYEK 360
361 GNFIGPTILS GVTPDMECYK EEIFGPVLVC MQANSFDEAI SIINKNKYGN GAAIFTSSGA 420
421 AARKFQMDIE AGQIGINVPI PVPLPFFSFT GNKASFAGDL NFYGKAGVDF FTQIKTVTQQ 480
481 WKDIPTSVSL AMPTSQKQ
|
| Detection Method: | |
| Confidence: | 145.0 |
| Match: | 1t90A |
| Description: | Crystal structure of methylmalonate semialdehyde dehydrogenase from Bacillus subtilis |
Matching Structure (courtesy of the PDB): |
|