| Protein: | gi|12597768, gi|... |
| Organism: | Arabidopsis thaliana |
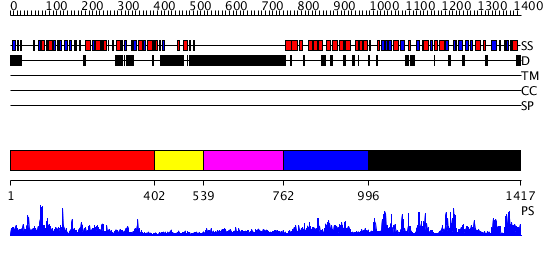
| Length: | 1417 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for gi|12597768, gi|....
| Description | E-value | Query Range |
Subject Range |
|
|
761.0 | [0..54] | [1413..43] |
|
|
686.0 | [0..460] | [1406..3] |
|
|
684.0 | [0..474] | [1406..1] |
|
|
681.0 | [0..740] | [1417..6] |
|
|
681.0 | [0..474] | [1413..1] |
|
|
680.0 | [0..474] | [1413..1] |
|
|
679.0 | [0..474] | [1413..1] |
|
Region A: Residues: [1-401] |
1 11 21 31 41 51
| | | | | |
1 MASDSAGATI SGNFSNSDNS ETLNLNTTKL YSSAISSISP QFPSPKPTSS CPSIPNSKRI 60
61 PNTNFIVDLF RLPHQSSSVA FFLSHFHSDH YSGLSSSWSK GIIYCSHKTA RLVAEILQVP 120
121 SQFVFALPMN QMVKIDGSEV VLIEANHCPG AVQFLFKVKL ESSGFEKYVH TGDFRFCDEM 180
181 RFDPFLNGFV GCDGVFLDTT YCNPKFVFPS QEESVGYVVS VIDKISEEKV LFLVATYVVG 240
241 KEKILVEIAR RCKRKIVVDA RKMSMLSVLG CGEEGMFTED ENESDVHVVG WNVLGETWPY 300
301 FRPNFVKMNE IMVEKGYDKV VGFVPTGWTY EVKRNKFAVR FKDSMEIHLV PYSEHSNYDE 360
361 LREFIKFLKP KRVIPTVGVD IEKFDCKEVN KMQKHFSGLV D
|
| Detection Method: | |
| Confidence: | 11.221849 |
| Match: | 1e5dA |
| Description: | Rubredoxin oxygen:oxidoreductase (ROO), C-terminal domain; Rubredoxin oxygen:oxidoreductase (ROO), N-terminal domain |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [402-538] |
1 11 21 31 41 51
| | | | | |
1 EMANKKDFLL GFYRQSYQKN EKSDVDVVSH SAEVYEEEEK NACEDGGENV PSSRGPILHD 60
61 TTPSSDSRLL IKLRDSLPAW VTEEQMLDLI KKHAGNPVDI VSNFYEYEAE LYKQASLPTP 120
121 SLNNQAVLFD DDVTDLQ
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [539-761] |
1 11 21 31 41 51
| | | | | |
1 PNPVKGICPD VQAIQKGFDL PRKMNLTKGT ISPGKRGKSS GSKSNKKAKK DPKSKPVGPG 60
61 QPTLFKFFNK VLDGGSNSVS VGSETEECNT DKKMVHIDAS EAYKEVTDQF IDIVNGSESL 120
121 RDYAASIIDE AKGDISRALN IYYSKPREIP GDHAGERGLS SKTIQYPKCS EACSSQEDKK 180
181 ASENSGHAVN ICVQTSAEES VDKNYVSLPP EKYQPKEHAC WRE
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [762-995] |
1 11 21 31 41 51
| | | | | |
1 GQPAPYIHLV RTFASVESEK GKIKAMSMLC NMFRSLFALS PEDVLPAVYL CTNKIAADHE 60
61 NIELNIGGSL ISSALEEACG ISRSTVRDMY NSLGDLGDVA QLCRQTQKLL VPPPPLLVRD 120
121 VFSTLRKISV QTGTGSTRLK KNLIVKLMRS CREKEIKFLV RTLARNLRIG AMLRTVLPAL 180
181 GRAIVMNSFW NDHNKELSES CFREKLEGVS AAVVEAYNIL PSLDVVVPSL MDKD
|
| Detection Method: | |
| Confidence: | 3.221849 |
| Match: | 1r7rA |
| Description: | The crystal structure of murine p97/VCP at 3.6A |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [996-1417] |
1 11 21 31 41 51
| | | | | |
1 IEFSTSTLSM VPGIPIKPML AKIAKGVQEF FNLSQEKAFT CEYKYDGQRA QIHKLLDGTV 60
61 CIFSRNGDET TSRFPDLVDV IKQFSCPAAE TFMLDAEVVA TDRINGNKLM SFQELSTRER 120
121 GSKDALITTE SIKVEVCVFV FDIMFVNGEQ LLALPLRERR RRLKEVFPET RPGYLEYAKE 180
181 ITVSATVGAE EASLNNHDTL SRINAFLEEA FQSSCEGIMV KSLDVNAGYC PTKRSDSWLK 240
241 VKRDYVDGLG DTLDLVPIGA WYGNGRKAGW YSPFLMACFN PETEEFQSVC RVMSGFSDAF 300
301 YIEVMKSSTL TLLFNDETDG MKEFYSEDKI LAKKPPYYRT GETPDMWFSA EVVWEIRGAD 360
361 FTVSPVHSAS LGLVHPSRGI SVRFPRFISK VTDRNPEECS TATDIAEMFH AQTRKMNITS 420
421 QH
|
| Detection Method: | |
| Confidence: | 33.522879 |
| Match: | 1a0iA |
| Description: | ATP-DEPENDENT DNA LIGASE FROM BACTERIOPHAGE T7 COMPLEX WITH ATP |
Matching Structure (courtesy of the PDB): |
|