| Protein: | STI_ARATH |
| Organism: | Arabidopsis thaliana |
| Length: | 1218 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for STI_ARATH.
| Description | E-value | Query Range |
Subject Range |
|
|
607.0 | [0..1] | [1211..1] |
|
|
494.0 | [0..336] | [1137..57] |
|
|
493.0 | [0..313] | [1137..33] |
|
|
492.0 | [0..313] | [1137..33] |
|
|
492.0 | [0..336] | [1143..57] |
|
|
492.0 | [0..336] | [1137..50] |
|
|
491.0 | [0..313] | [1137..29] |
|
|
491.0 | [0..336] | [1137..50] |
|
|
491.0 | [0..313] | [1137..29] |
|
|
490.0 | [0..313] | [1137..29] |
|
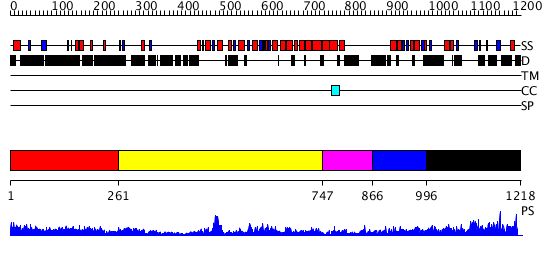
Region A: Residues: [1-260] |
1 11 21 31 41 51
| | | | | |
1 MSGSRVSDLS KLHLKKELTQ IRKAGRVLRD PGTTSSWKSP LDSSRSVALL ETPASRNGGS 60
61 SSQFPIRGES STNRRGKEKK VFLYNWKTQK SSSEKSGLAK NGKEEEEEEE DASSWTQASV 120
121 NDDDDVSDAR NGGDSYRREI QSASMGFRCR DTNLASQGVS KMRKSNVGSC KKKSKKKISS 180
181 SRLDCLSKYQ PRDDIVARNC NAGSDDTEEE LSNSEDLRKV TGASPLLLKL KQKNWSRSSS 240
241 RLLRANNRKE DSSCTYNSTP
|
| Detection Method: | |
| Confidence: | 72.09691 |
| Match: | 1ksfX |
| Description: | N-terminal, ClpS-binding domain of ClpA, an Hsp100 chaperone; ClpA, an Hsp100 chaperone, AAA+ modules |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [261-746] |
1 11 21 31 41 51
| | | | | |
1 ALSTSSYNMY AVRNPSTVGS WDGTTTSVND GDDELDDNLD LPGRQGCGIP CYWTKKAMKH 60
61 RGGCRSCCSP SFSDTLRRTG SSILCGSQSV YRRHNRHSSG GYSKQKIACR SAQGVLPLLS 120
121 YGGDGRGGSS LGTGLSDDEL STNYGELDLE AQSRLDGRRW STSYRSQDGL EAVALDGEEE 180
181 EGSTPETIRS FSQKYRPMFF EELIGQSIVV QSLMNAVKRS RIAPVYLFQG PRGTGKTSTA 240
241 RIFSAALNCV ATEEMKPCGY CKECNDFMSG KSKDFWELDG ANKKGADKVR YLLKNLPTIL 300
301 PRNSSMYKVF VIDECHLLPS KTWLSFLKFL ENPLQKVVFI FITTDLENVP RTIQSRCQKF 360
361 LFDKLKDSDI VVRLKKIASD ENLDVDLHAL DLIAMNADGS LRDAETMLEQ LSLLGKRITT 420
421 ALVNELVGVV SDEKLLELLE LALSSDTAET VKRARELLDL GADPIVLMSQ LASLIMDIIA 480
481 GTYKVV
|
| Detection Method: | |
| Confidence: | 37.39794 |
| Match: | 1e32A |
| Description: | Membrane fusion atpase p97 N-terminal domain , P97-Nn; Membrane fusion atpase p97, D1 domain; Membrane fusion atpase p97 domain 2, P97-Nc |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [747-865] |
1 11 21 31 41 51
| | | | | |
1 DEKYSNAFFD GRNLTEADME GLKHALKLLS EAEKQLRVSN DRSTWFTATL LQLGSMPSPG 60
61 TTHTGSSRRQ SSRATDDDPA SVSREVMAYK QRIGGLHFSK SASPASVIKR NGNHSHEAK
|
| Detection Method: | |
| Confidence: | 65.09691 |
| Match: | 1jr3A |
| Description: | gamma subunit; gamma subunit of DNA polymerase III, N-domain |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [866-995] |
1 11 21 31 41 51
| | | | | |
1 PFSRVIDNNC YKSSSSSQMI ESEGSIASHE NSIASTMMLN QRSSEKLNDI WRKCIERCHS 60
61 KTLRQLLYTH GKLISISEVE GILVAYIAFG ENDIKLRAER FLSSITNSIE MVLRRSVEVR 120
121 IILLPETELL
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [996-1218] |
1 11 21 31 41 51
| | | | | |
1 VVPHQTRKPE MTNKSGHLNN IAGLNAETDV EVGSSVESRS KLPMQRIESI IREQRLETAW 60
61 LQTADKDTPG SIIRVKPERN QILPQEDTYR QTNVASAISS SGLTTHQWVD ELNNEVKLLK 120
121 IGDNGELQEN LTGTRGQHCP LSPSLLHDTN FGNNKDNLGG YESGSGRVGC NILFCWNTKK 180
181 TQRRSKSKQV KGTPVRSRRN RKSRFSLFNG CAKPRKAEGN IRR
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.