| Protein: | PLDD1_ARATH |
| Organism: | Arabidopsis thaliana |
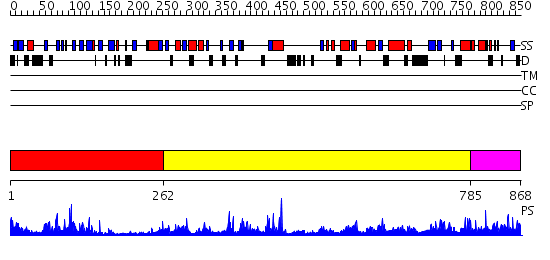
| Length: | 868 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for PLDD1_ARATH.
| Description | E-value | Query Range |
Subject Range |
|
|
974.0 | [0..1] | [868..1] |
|
Region A: Residues: [1-261] |
1 11 21 31 41 51
| | | | | |
1 MAEKVSEDVM LLHGDLDLKI VKARRLPNMD MFSEHLRRLF TACNACARPT DTDDVDPRDK 60
61 GEFGDKNIRS HRKVITSDPY VTVVVPQATL ARTRVLKNSQ EPLWDEKFNI SIAHPFAYLE 120
121 FQVKDDDVFG AQIIGTAKIP VRDIASGERI SGWFPVLGAS GKPPKAETAI FIDMKFTPFD 180
181 QIHSYRCGIA GDPERRGVRR TYFPVRKGSQ VRLYQDAHVM DGTLPAIGLD NGKVYEHGKC 240
241 WEDICYAISE AHHMIYIVGW S
|
| Detection Method: | |
| Confidence: | 28.09691 |
| Match: | 1dqvA |
| Description: | Synaptogamin I |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [262-784] |
1 11 21 31 41 51
| | | | | |
1 IFHKIKLVRE TKVPRDKDMT LGELLKYKSQ EGVRVLLLVW DDKTSHDKFG IKTPGVMGTH 60
61 DEETRKFFKH SSVICVLSPR YASSKLGLFK QQASPSSSIY IMTVVGTLFT HHQKCVLVDT 120
121 QAVGNNRKVT AFIGGLDLCD GRYDTPEHRI LHDLDTVFKD DFHNPTFPAG TKAPRQPWHD 180
181 LHCRIDGPAA YDVLINFEQR WRKATRWKEF SLRLKGKTHW QDDALIRIGR ISWILSPVFK 240
241 FLKDGTSIIP EDDPCVWVSK EDDPENWHVQ IFRSIDSGSV KGFPKYEDEA EAQHLECAKR 300
301 LVVDKSIQTA YIQTIRSAQH FIYIENQYFL GSSYAWPSYR DAGADNLIPM ELALKIVSKI 360
361 RAKERFAVYV VIPLWPEGDP KSGPVQEILY WQSQTMQMMY DVIAKELKAV QSDAHPLDYL 420
421 NFYCLGKREQ LPDDMPATNG SVVSDSYNFQ RFMIYVHAKG MIVDDEYVLM GSANINQRSM 480
481 AGTKDTEIAM GAYQPNHTWA HKGRHPRGQV YGYRMSLWAE HLG
|
| Detection Method: | |
| Confidence: | 42.30103 |
| Match: | 1v0rA |
| Description: | Tungstate-inhibited phospholipase D from Streptomyces sp. strain PMF. |
Matching Structure (courtesy of the PDB): |
|
| Term | Confidence | Notes |
| catalytic activity | 0.762303478549781 | bayes_pls_golite062009 |
|
Region A: Residues: [785-868] |
1 11 21 31 41 51
| | | | | |
1 KTGDEFVEPS DLECLKKVNT ISEENWKRFI DPKFSELQGH LIKYPLQVDV DGKVSPLPDY 60
61 ETFPDVGGKI IGAHSMALPD TLTT
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.