| Protein: | CLCF_ARATH |
| Organism: | Arabidopsis thaliana |
| Length: | 781 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for CLCF_ARATH.
| Description | E-value | Query Range |
Subject Range |
|
|
505.0 | [0..1] | [781..1] |
|
|
477.0 | [0..1] | [758..1] |
|
|
402.0 | [0..7] | [781..4] |
|
|
398.0 | [0..17] | [757..13] |
|
|
397.0 | [0..17] | [758..13] |
|
|
397.0 | [0..17] | [757..13] |
|
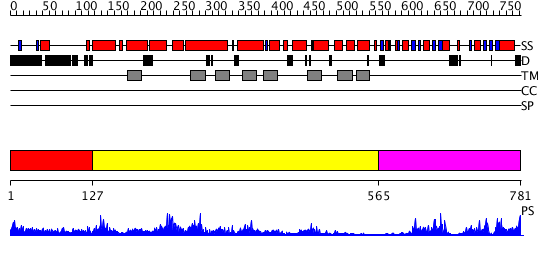
Region A: Residues: [1-126] |
1 11 21 31 41 51
| | | | | |
1 MSSGGAGEYN EDRHLLRSTD GDEVGIGGGE GDLDVESQSP AIRSGAGGVR DLFKHIDRRF 60
61 SLSGRRLSFK RMENIRVDRE RHNPSSSSAF SAAGEEDGGG ISNLHSVDDR NDEYGFDEEV 120
121 LGDSAP
|
| Detection Method: |
Shown below are all of our de novo (Rosetta) predictions for this domain.
Click here to view only most confident match.
| MCM Score |
SCOP Match |
SCOP Description | ||
| View | Download | 0.282 | a.45.1 | Glutathione S-transferase (GST), C-terminal domain |
| View | Download | 0.282 | a.45.1 | Glutathione S-transferase (GST), C-terminal domain |
|
Region A: Residues: [127-564] |
1 11 21 31 41 51
| | | | | |
1 PEWALLLIGC LIGVAAGICV AGFNKGVHVI HEWAWAGTPN EGAAWLRLQR LADTWHRILL 60
61 IPVTGGVIVG MMHGLLEILD QIRQSNSSQR QGLDFLAGIY PVIKAIQAAV TLGTGCSLGP 120
121 EGPSVDIGKS CANGFALMME NNRERRIALT AAGAASGIAS GFNAAVAGCF FAIETVLRPL 180
181 RAENSPPFTT AMIILASVIS STVSNALLGT QSAFTVPSYD LKSAAELPLY LILGMLCGAV 240
241 SVVFSRLVTW FTKSFDFIKD KFGLPAIVCP ALGGLGAGII ALKYPGILYW GFTNVEEILH 300
301 TGKSASAPGI WLLAQLAAAK VVATALCKGS GLVGGLYAPS LMIGAAVGAV FGGSAAEIIN 360
361 RAIPGNAAVA QPQAYALVGM AATLASMCSV PLTSVLLLFE LTKDYRILLP LMGAVGLAIW 420
421 VPSVANQGKE SDSSEGRS
|
| Detection Method: | |
| Confidence: | 59.522879 |
| Match: | 1kplA |
| Description: | Clc chloride channel |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [565-781] |
1 11 21 31 41 51
| | | | | |
1 TGRGYSSLSP SERKTEGVWR HTDNADSLEL TVIENPDHNS FLDEETILED LKVMRVMSKN 60
61 YVKVSSGTTL REARNILKES HQNCIMVVDD DDFLAGILTH GDIRRYLSNN ASTILDENTC 120
121 PVSSVCTKKI SYRGQERGLL TCYPDATVGV AKELMEARGV KQLPVVKRGE VIHKGKRRKL 180
181 LGLLHYDSIW TFLRDEMSRR RSINDRRKDK EVGTNGH
|
| Detection Method: | |
| Confidence: | 31.522879 |
| Match: | 1vrdA |
| Description: | Crystal structure of Inosine-5'-monophosphate dehydrogenase (TM1347) from THERMOTOGA MARITIMA at 2.18 A resolution |
Matching Structure (courtesy of the PDB): |
|