| Protein: | fma1 |
| Organism: | Schizosaccharomyces pombe |
| Length: | 379 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for fma1.
| Description | E-value | Query Range |
Subject Range |
|
|
399.0 | [0..1] | [378..1] |
|
|
387.0 | [0..1] | [378..1] |
|
|
384.0 | [0..1] | [370..79] |
|
|
382.0 | [0..1] | [378..1] |
|
|
381.0 | [0..1] | [370..1] |
|
|
381.0 | [0..1] | [378..1] |
|
|
379.0 | [0..1] | [370..9] |
|
|
377.0 | [0..1] | [370..1] |
|
|
377.0 | [0..1] | [370..1] |
|
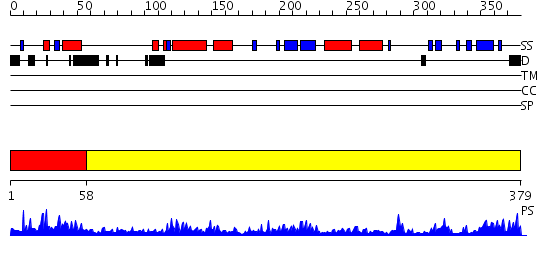
Region A: Residues: [1-57] |
1 11 21 31 41 51
| | | | | |
1 MATEIAKHIC CGIDCNNEAD RLQCPKCLND GVKSYFCGQE CFRNSWNIHK HLHRPPN
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
| MCM Score |
GO Score |
GO Term |
SCOP Match |
SCOP Description | ||
| View | Download | 0.537 | N/A | N/A | c.47.1 | Thioredoxin-like |
|
Region A: Residues: [58-379] |
1 11 21 31 41 51
| | | | | |
1 VEKREDGTYN PFPKFHFAGS LKPVYPLSPI RKVPPHIKRP DYAKTGVSRS EQIEGRSFKL 60
61 KRLTPKEQEG MRKVCRLGRE VLDAAAAAVR PGTTTDELDS IVHNACIERD CFPSTLNYYA 120
121 FPKSVCTSVN EIICHGIPDQ RPLEDGDIVN IDVSLYHNGF HGDLNETYYV GDKAKANPDL 180
181 VCLVENTRIA LDKAIAAVKP GVLFQEFGNI IEKHTNSITE KQISVVRTYC GHGINQLFHC 240
241 SPSIPHYSHN KAPGIARPGM TFTIEPMLTL GPARDITWPD DWTSSTASGR CSAQFEHTLL 300
301 VTETGCEVLT ARLPNSPGGP LK
|
| Detection Method: | |
| Confidence: | 79.69897 |
| Match: | 2b3hA |
| Description: | Crystal structure of Human Methionine Aminopeptidase Type I with a third cobalt in the active site |
Matching Structure (courtesy of the PDB): |
|