| Protein: | g-PB |
| Organism: | Drosophila melanogaster |
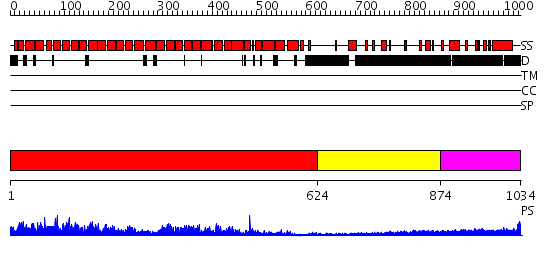
| Length: | 1034 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for g-PB.
| Description | E-value | Query Range |
Subject Range |
|
|
640.0 | [0..1] | [1034..1] |
|
Region A: Residues: [1-623] |
1 11 21 31 41 51
| | | | | |
1 MALKKVKGNF FERMFDKNLT DLVRGIRNNK DNEAKYISTC IEEIKQELRQ DNISVKCNAV 60
61 AKLTYIQMLG YDISWAGFNI IEVMSSSRFT CKRIGYLAAS QCFHPDSELL MLTTNMIRKD 120
121 LNSQNQYDAG VALSGLSCFI SPDLSRDLAN DIMTLMSSTK PYLRMKAVLM MYKVFLRYPE 180
181 ALRPAFPKLK EKLEDPDPGV QSAAVNVICE LARKNPKNYL PLAPIFFKLM TTSTNNWMLI 240
241 KIIKLFGALT PLEPRLGKKL IEPLTNLIHS TSAMSLLYEC INTVIAVLIS ISSGMPNHSA 300
301 SIQLCVQKLR ILIEDSDQNL KYLGLLAMSK ILKTHPKSVQ AHKDLILACL DDKDESIRLR 360
361 ALDLLYGMVS KKNLMEIVKR LLGHMERAEG SAYRDELLYK VIEICAQSSY LYVTNFEWYL 420
421 TVLVELIQLE AGSRHGRLIA EQLLDVAIRV PVVRQFAVNE MTNLLDTFTV SAQSNSMYEV 480
481 LYAAAWIVGE FAGELEDAEK TLNILLRPRL LPGHIQGVYV QNVIKLFARL ATTCLELQDL 540
541 PGLVTLCDHV LDKLQHFNGS SDIEVQERAN SACMLIEMLR NQLSTSTDAM AMDTTTEGGI 600
601 PPLAIEIVQE MTLLFTGELI PVA
|
| Detection Method: | |
| Confidence: | 129.0 |
| Match: | 1w63A |
| Description: | AP1 clathrin adaptor core |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [624-873] |
1 11 21 31 41 51
| | | | | |
1 PKAQRKVPLP DGLDLDEWIN APPPEDAASS SSSEHDKDEL FVSATQAGTG ADGGEKRRQS 60
61 LELTPEQLER QRMARLIEQS NNPHYLKSTP TASGASNADQ YDNIDDIPIT ELPLDMEGVA 120
121 ALRVGITKRS DKYLQEQQAA QGSKDGKKKH KKGKKSKKAK NKVAYNSSSE SEGEPKPLHI 180
181 VNTTLDMPEG VSMSDSEDKD GKYDPNDPHR ALDIELDITE FEAPAVRSAS KKSAADKENL 240
241 KTPADSAGGG
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [874-1034] |
1 11 21 31 41 51
| | | | | |
1 NAAKKDRKKD KDKDKERKVK REHRESKRER KEAAVQSVID LIDADTPTPS PSHISATSNN 60
61 NNTSTVLPDA PKHHKKKKNK EKTTDEAPDA LATATGSSII DVGGEEASEV ASKVHKKKHK 120
121 KEKSQRKEKK KASESASVSA IVSIGDYEQP LGISTPSKEI L
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.