| Protein: | CHR25_ARATH |
| Organism: | Arabidopsis thaliana |
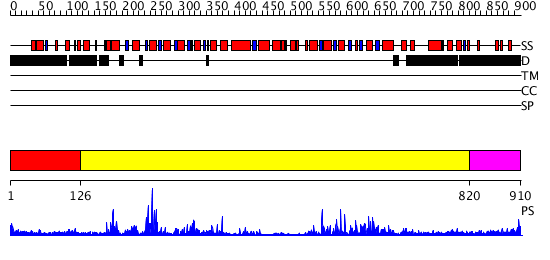
| Length: | 910 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
No multiple sequence alignment data found for CHR25_ARATH.
|
Region A: Residues: [1-125] |
1 11 21 31 41 51
| | | | | |
1 MEEEDEEILS SSDCDDSSDS YKDDSQDSEG ENDNPECEDL AVVSLSSDAD RKSKNVKDLL 60
61 RGNLVVQRQP LLPRVLSVSD GAAVCRKPFK PPCSHGYDST GQLSRRLSAR KRFVPWGSST 120
121 PVVVA
|
| Detection Method: | |
| Confidence: | 9.0 |
| Match: | 2b2uA |
| Description: | Tandem chromodomains of human CHD1 complexed with Histone H3 Tail containing trimethyllysine 4 and dimethylarginine 2 |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [126-819] |
1 11 21 31 41 51
| | | | | |
1 LPTKLEASTN IERDEEEEVV CLPPDIEPLV LWQSEEDGMS NVTTIMVHSV LVKFLRPHQR 60
61 EGVQFMFDCV SGLHGSANIN GCILADDMGL GKTLQSITLL YTLLCQGFDG TPMVKKAIIV 120
121 TPTSLVSNWE AEIKKWVGDR IQLIALCEST RDDVLSGIDS FTRPRSALQV LIISYETFRM 180
181 HSSKFCQSES CDLLICDEAH RLKNDQTLTN RALASLTCKR RVLLSGTPMQ NDLEEFFAMV 240
241 NFTNPGSLGD AAHFRHYYEA PIICGREPTA TEEEKNLAAD RSAELSSKVN QFILRRTNAL 300
301 LSNHLPPKII EVVCCKMTTL QSTLYNHFIS SKNLKRALAD NAKQTKVLAY ITALKKLCNH 360
361 PKLIYDTIKS GNPGTVGFEN CLEFFPAEMF SGRSGAWTGG DGAWVELSGK MHVLSRLLAN 420
421 LRRKTDDRIV LVSNYTQTLD LFAQLCRERR YPFLRLDGST TISKRQKLVN RLNDPTKDEF 480
481 AFLLSSKAGG CGLNLIGANR LVLFDPDWNP ANDKQAAARV WRDGQKKRVY VYRFLSTGTI 540
541 EEKVYQRQMS KEGLQKVIQH EQTDNSTRQG NLLSTEDLRD LFSFHGDVRS EIHEKMSCSR 600
601 CQNDASGTEN IEEGNENNVD DNACQIDQED IGGFAKDAGC FNLLKNSERQ VGTPLEEDLG 660
661 SWGHHFTSKS VPDAILQASA GDEVTFVFTN QVDG
|
| Detection Method: | |
| Confidence: | 104.0 |
| Match: | 1z3iX |
| Description: | Structure of the SWI2/SNF2 chromatin remodeling domain of eukaryotic Rad54 |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [820-910] |
1 11 21 31 41 51
| | | | | |
1 KLVPIESNVS PKTVESEEHN RNQPVNKRAF NKPQQRPREP LQPLSLNETT KRVKLSTYKR 60
61 LHGNSNIDDA QIKMSLQRPN LVSVNHDDDF V
|
| Detection Method: | |
| Confidence: | 4.0 |
| Match: | 1tf2A |
| Description: | Crystal structure of SecA:ADP in an open conformation from Bacillus Subtilis |
Matching Structure (courtesy of the PDB): |
|