| Protein: | HSF1 |
| Organism: | Saccharomyces cerevisiae |
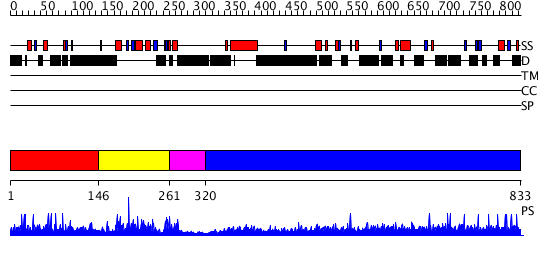
| Length: | 833 amino acids |
| Reference: | Malmström L, et al. (2007) Superfamily assignments for the yeast proteome through integration of structure prediction with the gene ontology. PLoS Biol 5(4): e76. doi:10.1371/journal.pbio.0050076 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for HSF1.
| Description | E-value | Query Range |
Subject Range |
|
|
994.0 | [0..1] | [833..1] |
|
|
439.0 | [0..119] | [728..6] |
|
|
435.0 | [0..119] | [728..6] |
|
|
310.0 | [0..48] | [422..53] |
|
|
293.0 | [0..124] | [506..13] |
|
|
270.0 | [0..137] | [488..2] |
|
|
268.0 | [0..161] | [497..4] |
|
|
265.0 | [0..153] | [514..10] |
|
Region A: Residues: [1-145] |
1 11 21 31 41 51
| | | | | |
1 MNNAANTGTT NESNVSDAPR IEPLPSLNDD DIEKILQPND IFTTDRTDAS TTSSTAIEDI 60
61 INPSLDPQSA ASPVPSSSFF HDSRKPSTST HLVRRGTPLG IYQTNLYGHN SRENTNPNST 120
121 LLSSKLLAHP PVPYGQNPDL LQHAV
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [146-260] |
1 11 21 31 41 51
| | | | | |
1 YRAQPSSGTT NAQPRQTTRR YQSHKSRPAF VNKLWSMLND DSNTKLIQWA EDGKSFIVTN 60
61 REEFVHQILP KYFKHSNFAS FVRQLNMYGW HKVQDVKSGS IQSSSDDKWQ FENEN
|
| Detection Method: | |
| Confidence: | 363.218487 |
| Match: | 3htsB |
| Description: | Heat-shock transcription factor |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [261-319] |
1 11 21 31 41 51
| | | | | |
1 FIRGREDLLE KIIRQKGSSN NHNSPSGNGN PANGSNIPLD NAAGSNNSNN NISSSNSFF
|
| Detection Method: | |
| Confidence: | 9.77 |
| Match: | |
| Description: | No description for 3htsB was found. |
|
Region A: Residues: [320-833] |
1 11 21 31 41 51
| | | | | |
1 NNGHLLQGKT LRLMNEANLG DKNDVTAILG ELEQIKYNQI AISKDLLRIN KDNELLWQEN 60
61 MMARERHRTQ QQALEKMFRF LTSIVPHLDP KMIMDGLGDP KVNNEKLNSA NNIGLNRDNT 120
121 GTIDELKSND SFINDDRNSF TNATTNARNN MSPNNDDNSI DTASTNTTNR KKNIDENIKN 180
181 NNDIINDIIF NTNLANNLSN YNSNNNAGSP IRPYKQRYLL KNRANSSTSS ENPSLTPFDI 240
241 ESNNDRKISE IPFDDEEEEE TDFRPFTSRD PNNQTSENTF DPNRFTMLSD DDLKKDSHTN 300
301 DNKHNESDLF WDNVHRNIDE QDARLQNLEN MVHILSPGYP NKSFNNKTSS TNTNSNMESA 360
361 VNVNSPGFNL QDYLTGESNS PNSVHSVPSN GSGSTPLPMP NDNDTEHAST SVNQGENGSG 420
421 LTPFLTVDDH TLNDNNTSEG STRVSPDIKF SATENTKVSD NLPSFNDHSY STQADTAPEN 480
481 AKKRFVEEIP EPAIVEIQDP TEYNDHRLPK RAKK
|
| Detection Method: | |
| Confidence: | 7.24 |
| Match: | |
| Description: | No description for 1i3qA was found. |