| Protein: | RTP1_YEAST |
| Organism: | Saccharomyces cerevisiae S288c |
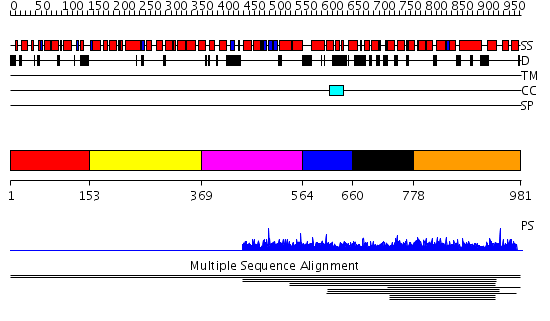
| Length: | 981 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for RTP1_YEAST.
| Description | E-value | Query Range |
Subject Range |
|
|
0.0 | [1..981] | [1..981] |
|
|
0.0 | [448..936] | [318..816] |
|
|
0.0 | [448..936] | [313..811] |
|
|
0.0 | [538..934] | [388..744] |
|
|
2.0E-81 | [608..975] | [422..735] |
|
|
1.0E-60 | [730..934] | [841..1031] |
|
Region A: Residues: [1-152] |
1 11 21 31 41 51
| | | | | |
1 MNEDKEQKIN IHDILNTRPK LTKKTALDVF FEDLDDNVIT PINEYVLDSG SSSSSSIYQA 60
61 LKCSNNNEFV AVLLQKFQNL HIHVLEQQRR LIESKSDLLP ISLHDMKYVD ELINLLIIHG 120
121 IDANLSPTMK IPFDSKRINT FKKGQKSAEY ET
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [153-368] |
1 11 21 31 41 51
| | | | | |
1 PRWHTINNDT LSQVITVFYN VLTSERSSDY LREIISKGSA YANILLGLIV LHLQLPNRYS 60
61 SQMITNLEDT QETYTLFGVY TLLVETIQDE KVREPILSKL TTLTLRRPEN GLISLIDFVL 120
121 GVRDAEDIDI EKFNRIYQIL MSKPKTMTNL QYLTELFKQI YDGLTFVNRP ILVTCLNGLI 180
181 LKFYLRNKRI VNDFLFKKVR SIIFNSPLTD HTAKEL
|
| Detection Method: | |
| Confidence: | 13.5 |
| Match: | 1qgkA |
| Description: | Importin beta |
Matching Structure (courtesy of the PDB): |
|
| Term | Confidence | Notes |
| binding | 1.4666436207573 | bayes_pls_golite062009 |
| protein binding | 1.12452075263773 | bayes_pls_golite062009 |
| nucleic acid binding | 0.410251595672781 | bayes_pls_golite062009 |
|
Region A: Residues: [369-563] |
1 11 21 31 41 51
| | | | | |
1 NDVINVLISL SKNSSSDLLN DLVTSCPDED GTTPGQFFLY VWIYALFLKK NQKLDPLEIN 60
61 KLSISDNKST DSIHFPEQSS SKYYEVVLSL LKSLIVITEN FQYLNVLSLN LLNFEHEKWK 120
121 YLIDLDTQLP YISVKNTDMA ELFFEKGSKN SQISEFLQDM DLSIELFMEF LVLLNDEEQS 180
181 KTLFLDILKR WVHHT
|
| Detection Method: | |
| Confidence: | 13.5 |
| Match: | 1qgkA |
| Description: | Importin beta |
| Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [564-659] |
1 11 21 31 41 51
| | | | | |
1 KKSEKRSSDN HSGMPSVTDN ALILMDLKLL ECMNNRFKTK IVNKPKDVLI VIDQLIDVVQ 60
61 EKDETIQEVE ADSDDEVEEG EETEELDPNE NSSYKI
|
| Detection Method: | |
| Confidence: | 13.5 |
| Match: | 1qgkA |
| Description: | Importin beta |
| Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [660-777] |
1 11 21 31 41 51
| | | | | |
1 ILQLLSTVLS ESSSSILLQN SYILKSISRK LQSFNTNASE IDALLASIDN ILINGHTTER 60
61 NDNIEIEMDE ERLDKAITSL HDPLVPIKSY GLTELRHLAE KKSPVISLEK VLQIHLDY
|
| Detection Method: | |
| Confidence: | 13.5 |
| Match: | 1qgkA |
| Description: | Importin beta |
| Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [778-981] |
1 11 21 31 41 51
| | | | | |
1 LKNMDPFIYL NVIKGLTTLC ELEPETILPL LAEFYANKKK KNRLDDVLKV GEVFINYIQR 60
61 QNELFQGKLA YLIIDTCLSI VRPNDSKPLD NRWRMSSMSI LGMCLQINAR GVSDRIRDML 120
121 DCVFGILQLE QPQNHLKDKD DSFLMRRSAV HLIHDLLYST GFDLLPFEYN YDKLKTLLSY 180
181 VRDQDEDYMV CEQIDKLLTV LDSL
|
| Detection Method: | |
| Confidence: | 13.5 |
| Match: | 1qgkA |
| Description: | Importin beta |
| Matching Structure (courtesy of the PDB): |
|