| Protein: | TOM71_YEAST |
| Organism: | Saccharomyces cerevisiae S288c |
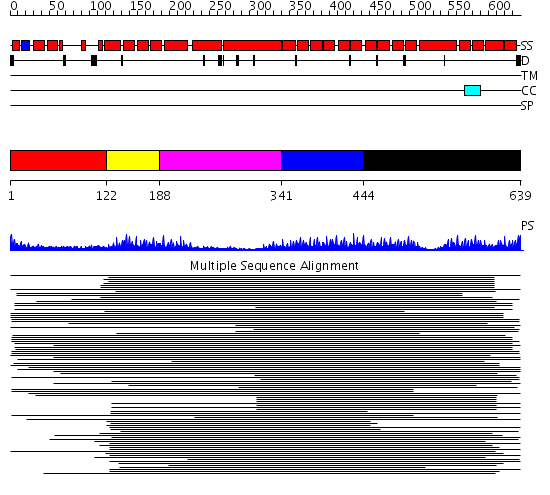
| Length: | 639 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for TOM71_YEAST.
| Description | E-value | Query Range |
Subject Range |
|
|
1.0E-84 | [123..607] | [7..462] |
|
|
2.0E-84 | [117..607] | [11..472] |
|
|
2.0E-84 | [123..607] | [7..462] |
|
|
3.0E-82 | [113..607] | [36..501] |
|
|
5.0E-79 | [1..639] | [1..639] |
|
|
1.0E-77 | [8..567] | [67..617] |
|
|
3.0E-76 | [113..604] | [39..501] |
|
|
1.0E-66 | [77..638] | [17..559] |
|
Region A: Residues: [1-121] |
1 11 21 31 41 51
| | | | | |
1 MAENSLLRFI TKNKVAILAT VSAGTAAVGA YVYYQQIKQQ QQQQLKGTKD NRRQSEAFAG 60
61 QNEDEADLKD DGSVVSGSNK RKKKKNKRKR NNKAKSGEGF DYPSLPNGEP DIAQLKGLSP 120
121 S
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [122-187] |
1 11 21 31 41 51
| | | | | |
1 QRQAYAVQLK NRGNHFFTAK NFNEAIKYYQ YAIELDPNEP VFYSNISACY ISTGDLEKVI 60
61 EFTTKA
|
| Detection Method: | |
| Confidence: | 114.594777 |
| Match: | 1a17__ |
| Description: | Protein phosphatase 5 |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [188-340] |
1 11 21 31 41 51
| | | | | |
1 LEIKPDHSKA LLRRASANES LGNFTDAMFD LSVLSLNGDF DGASIEPMLE RNLNKQAMKV 60
61 LNENLSKDEG RGSQVLPSNT SLASFFGIFD SHLEVSSVNT SSNYDTAYAL LSDALQRLYS 120
121 ATDEGYLVAN DLLTKSTDMY HSLLSANTVD DPL
|
| Detection Method: | |
| Confidence: | 9.85 |
| Match: | 1jcqA |
| Description: | Protein farnesyltransferase alpha-subunit |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [341-443] |
1 11 21 31 41 51
| | | | | |
1 RENAALALCY TGIFHFLKNN LLDAQVLLQE SINLHPTPNS YIFLALTLAD KENSQEFFKF 60
61 FQKAVDLNPE YPPTYYHRGQ MYFILQDYKN AKEDFQKAQS LNP
|
| Detection Method: | |
| Confidence: | 59.70927 |
| Match: | 1elrA_ |
| Description: | Hop |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [444-639] |
1 11 21 31 41 51
| | | | | |
1 ENVYPYIQLA CLLYKQGKFT ESEAFFNETK LKFPTLPEVP TFFAEILTDR GDFDTAIKQY 60
61 DIAKRLEEVQ EKIHVGIGPL IGKATILARQ SSQDPTQLDE EKFNAAIKLL TKACELDPRS 120
121 EQAKIGLAQL KLQMEKIDEA IELFEDSAIL ARTMDEKLQA TTFAEAAKIQ KRLRADPIIS 180
181 AKMELTLARY RAKGML
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
| MCM Score |
GO Score |
GO Term |
SCOP Match |
SCOP Description | ||
| View | Download | 0.874 | 0.276 | mitochondrial outer membrane | a.118.8 | TPR-like |