| Protein: | PLPD2_ARATH |
| Organism: | Arabidopsis thaliana |
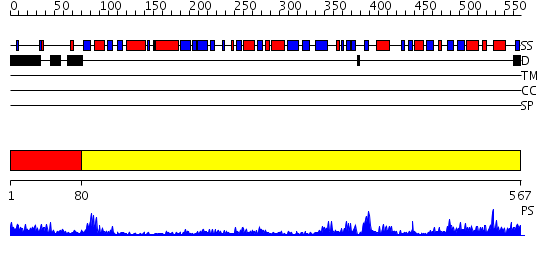
| Length: | 567 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for PLPD2_ARATH.
| Description | E-value | Query Range |
Subject Range |
|
|
385.0 | [0..82] | [557..2] |
|
Region A: Residues: [1-79] |
1 11 21 31 41 51
| | | | | |
1 MQSVLSLSFS QASLPLANRT LCSSNAAPST PRNLRFCGLR REAFCFSPSK QLTSCRFHIQ 60
61 SRRIEVSAAA SSSAGNGAP
|
| Detection Method: | |
| Confidence: | 18.69897 |
| Match: | 1djnA |
| Description: | Trimethylamine dehydrogenase, N-terminal domain; Trimethylamine dehydrogenase, C-terminal domain; Trimethylamine dehydrogenase, middle domain |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [80-567] |
1 11 21 31 41 51
| | | | | |
1 SKSFDYDLII IGAGVGGHGA ALHAVEKGLK TAIIEGDVVG GTCVNRGCVP SKALLAVSGR 60
61 MRELQNEHHM KAFGLQVSAA GYDRQGVADH ASNLATKIRN NLTNSMKALG VDILTGFGAV 120
121 LGPQKVKYGD NIITGKDIII ATGSVPFVPK GIEVDGKTVI TSDHALKLES VPDWIAIVGS 180
181 GYIGLEFSDV YTALGSEVTF IEALDQLMPG FDPEISKLAQ RVLINTRKID YHTGVFASKI 240
241 TPAKDGKPVL IELIDAKTKE PKDTLEVDAA LIATGRAPFT NGLGLENINV TTQRGFIPVD 300
301 ERMRVIDGNG KLVPHLYCIG DANGKLMLAH AASAQGISVV EQVTGRDHVL NHLSIPAACF 360
361 THPEISMVGL TEPQAREKAE KEGFKVSIAK TSFKANTKAL AENEGEGLAK MIYRPDNGEI 420
421 LGVHIFGLHA ADLIHEASNA IALGTRIQDI KLAVHAHPTL SEVVDELFKA AKVDSPASVT 480
481 AQSVKVTV
|
| Detection Method: | |
| Confidence: | 103.0 |
| Match: | 1zy8A |
| Description: | The crystal structure of dihydrolipoamide dehydrogenase and dihydrolipoamide dehydrogenase-binding protein (didomain) subcomplex of human pyruvate dehydrogenase complex. |
Matching Structure (courtesy of the PDB): |
|