| Protein: | gi|208965416 |
| Organism: | synthetic construct |
| Length: | 631 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for gi|208965416.
| Description | E-value | Query Range |
Subject Range |
|
|
0.0 | [1..631] | [5..635] |
|
|
0.0 | [1..631] | [1..631] |
|
|
0.0 | [1..631] | [1..631] |
|
|
0.0 | [1..631] | [1..635] |
|
|
0.0 | [1..631] | [1..635] |
|
|
0.0 | [1..631] | [1..630] |
|
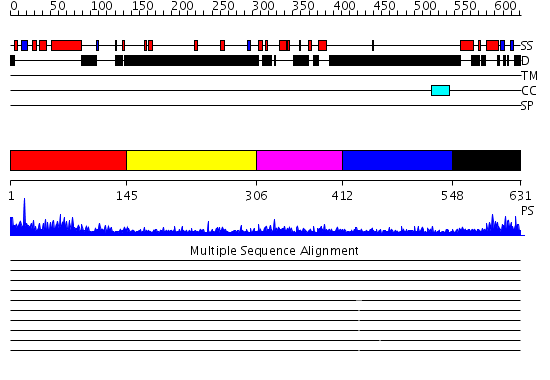
Region A: Residues: [1-144] |
1 11 21 31 41 51
| | | | | |
1 MFYAHFVLSK RGPLAKIWLA AHWDKKLTKA HVFECNLESS VESIISPKVK MALRTSGHLL 60
61 LGVVRIYHRK AKYLLADCNE AFIKIKMAFR PGVVDLPEEN REAAYNAITL PEEFHDFDQP 120
121 LPDLDDIDVA QQFSLNQSRV EEIT
|
| Detection Method: | |
| Confidence: | 67.920819 |
| Match: | PF04825.4 |
| Description: | No description for PF04825.4 was found. |
Shown below are all of our structure predictions for this domain.
Click here to view only most confident match.
| MCM Score |
GO Score |
GO Term |
SCOP Match |
SCOP Description | ||
| View | Download | 0.433 | 0.014 | double-strand break repair | a.7.1 | Spectrin repeat |
| View | Download | 0.472 | 0.013 | double-strand break repair | a.47.2 | t-snare proteins |
| View | Download | 0.574 | 0.005 | double-strand break repair | a.7.5 | Tubulin chaperone cofactor A |
| View | Download | 0.441 | 0.002 | double-strand break repair | f.36.1 | Neurotransmitter-gated ion-channel pransmembrane pore |
| View | Download | 0.434 | N/A | N/A | f.36.1 | Neurotransmitter-gated ion-channel pransmembrane pore |
|
Region A: Residues: [145-305] |
1 11 21 31 41 51
| | | | | |
1 MREEVGNISI LQENDFGDFG MDDREIMREG SAFEDDDMLV STTTSNLLLE SEQSTSNLNE 60
61 KINHLEYEDQ YKDDNFGEGN DGGILDDKLI SNNDGGIFDD PPALSEAGVM LPEQPAHDDM 120
121 DEDDNVSMGG PDSPDSVDPV EPMPTMTDQT TLVPNEEEAF A
|
| Detection Method: |
Shown below are all of our structure predictions for this domain.
Click here to view only most confident match.
Found no structure predictions for this domain.
|
Region A: Residues: [306-411] |
1 11 21 31 41 51
| | | | | |
1 LEPIDITVKE TKAKRKRKLI VDSVKELDSK TIRAQLSDYS DIVTTLDLAP PTKKLMMWKE 60
61 TGGVEKLFSL PAQPLWNNRL LKLFTRCLTP LVPEDLRKRR KGGEAD
|
| Detection Method: |
Shown below are all of our structure predictions for this domain.
Click here to view only most confident match.
| MCM Score |
GO Score |
GO Term |
SCOP Match |
SCOP Description | ||
| View | Download | 0.652 | 0.041 | double-strand break repair | a.35.1 | lambda repressor-like DNA-binding domains |
| View | Download | 0.482 | 0.012 | double-strand break repair | d.58.8 | Viral DNA-binding domain |
| View | Download | 0.765 | 0.007 | double-strand break repair | a.28.2 | Colicin E immunity proteins |
| View | Download | 0.540 | 0.006 | double-strand break repair | a.8.3 | Families 57/38 glycoside transferase middle domain |
| View | Download | 0.467 | 0.003 | double-strand break repair | d.52.1 | Alpha-lytic protease prodomain |
|
Region A: Residues: [412-547] |
1 11 21 31 41 51
| | | | | |
1 NLDEFLKEFE NPEVPREDQQ QQHQQRDVID EPIIEEPSRL QESVMEASRT NIDESAMPPP 60
61 PPQGVKRKAG QIDPEPVMPP QQVEQMEIPP VELPPEEPPN ICQLIPELEL LPEKEKEKEK 120
121 EKEDDEEEED EDASGG
|
| Detection Method: |
Shown below are all of our de novo (Rosetta) predictions for this domain.
Click here to view only most confident match.
Found no structure predictions for this domain.
|
Region A: Residues: [548-631] |
1 11 21 31 41 51
| | | | | |
1 DQDQEERRWN KRTQQMLHGL QRALAKTGAE SISLLELCRN TNRKQAAAKF YSFLVLKKQQ 60
61 AIELTQEEPY SDIIATPGPR FHII
|
| Detection Method: | |
| Confidence: | 6.0 |
| Match: | 1w1wE |
| Description: | Sc Smc1hd:Scc1-C complex, ATPgS |
Matching Structure (courtesy of the PDB): |
|
| Term | Confidence | Notes |
| transcription regulator activity | 2.18423275686836 | bayes_pls_golite062009 |
| DNA binding | 2.00689495715419 | bayes_pls_golite062009 |
| nucleic acid binding | 1.95069166062856 | bayes_pls_golite062009 |
| binding | 1.5962792529649 | bayes_pls_golite062009 |
| transcription factor activity | 1.3352343070618 | bayes_pls_golite062009 |