| Protein: | CSK_ARATH |
| Organism: | Arabidopsis thaliana |
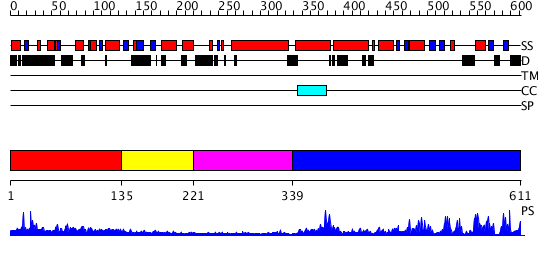
| Length: | 611 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for CSK_ARATH.
| Description | E-value | Query Range |
Subject Range |
|
|
356.0 | [0..1] | [611..1] |
|
|
268.0 | [0..241] | [607..6] |
|
|
262.0 | [0..59] | [609..248] |
|
|
260.0 | [0..89] | [610..209] |
|
|
260.0 | [0..68] | [610..759] |
|
|
260.0 | [0..332] | [603..467] |
|
|
258.0 | [0..228] | [601..344] |
|
Region A: Residues: [1-134] |
1 11 21 31 41 51
| | | | | |
1 MLLSAIASQT LLSSNPNLHF SNSIPNPRPS NPSLKLLNAS SSSSSSSSSS IFTRGLRYVN 60
61 HTVSNEESEP GGGETMVASA SAIASAIRGA STTPVEFTQM IEKDHLKTKI ILPSPDFQRL 120
121 CLEQLDLFRQ IVDP
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [135-220] |
1 11 21 31 41 51
| | | | | |
1 NAVLSIYVRP AGSYVMDRLE LRRVTCYPSV NAGDVVILVG NFGIPAGLRA AEASLSSQQV 60
61 ELVSKHRAAV FPMVKHPFVV GFLVAE
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [221-338] |
1 11 21 31 41 51
| | | | | |
1 LPVEAEEEEE EEEEEKPHGV NQFLSPEEAY ALPASANTKS PRVKLPSVKV FTEEQRSYAI 60
61 NISRTLAMAY VMDQKTMLLQ QSSWQNNVRM SKLVEQIRGP LSTMRTLSKM LSTHTKRN
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
| MCM Score |
SCOP Match |
SCOP Description | ||
| View | Download | 0.971 | a.47.2 | t-snare proteins |
|
Region A: Residues: [339-611] |
1 11 21 31 41 51
| | | | | |
1 QISHDIVEDL IVQGDQIKDT LEELQDAVHL TKANIVRHNE EALKKINKTH NETRRSKYEH 60
61 KDPIDGSQIS STRLSLGSGL DDSEMPMPPL ALAPLQMHSI RPCDISNVLL DMVETVRPLA 120
121 LTQQRVVELG ENSASLQVAV EEPALRQALS NLIEGALLRT HVGGKVEILS TRAPAGGSLV 180
181 VIDDDGPDMR YMTQMHSLTP FGAELLSENM VEDNMTWNFV AGLTVAREIL ESYGCVIRVI 240
241 SPRSSDAALG AGGTRVELWL PAFPAAVSEA NEA
|
| Detection Method: | |
| Confidence: | 53.69897 |
| Match: | 2c2aA |
| Description: | Structure of the entire cytoplasmic portion of a sensor histidine kinase protein |
Matching Structure (courtesy of the PDB): |
|