| Protein: | gi|42570548 |
| Organism: | Arabidopsis thaliana |
| Length: | 488 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for gi|42570548.
| Description | E-value | Query Range |
Subject Range |
|
|
432.0 | [0..37] | [488..496] |
|
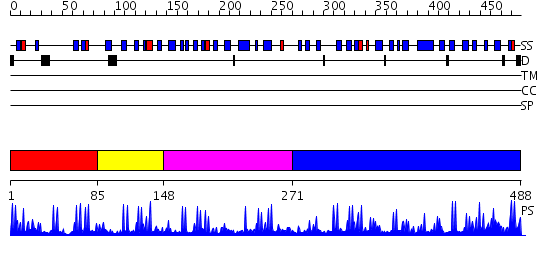
Region A: Residues: [1-84] |
1 11 21 31 41 51
| | | | | |
1 MSSTDDFVAD TTPELMFPFY HHPLSAYDYS SSYILQEKAY CCFMCVYSLF DKEPMLLPDI 60
61 DYHCTECGLN MHKKCIKRLF LNWP
|
| Detection Method: | |
| Confidence: | 1.15 |
| Match: | 1r79A |
| Description: | Solution Structure of The C1 Domain of The Human Diacylglycerol Kinase Delta |
Matching Structure (courtesy of the PDB): |
|
| Term | Confidence | Notes |
| binding | 0.0196698141692984 | bayes_pls_golite062009 |
|
Region A: Residues: [85-147] |
1 11 21 31 41 51
| | | | | |
1 FLCNHVLNIF QNTFTSSDNI DDFSCHFCQS RIKSSFARFT LCNINVDEKC MVELQPPLKI 60
61 SEP
|
| Detection Method: | |
| Confidence: | 5.522879 |
| Match: | 1tbnA |
| Description: | NMR STRUCTURE OF A PROTEIN KINASE C-G PHORBOL-BINDING DOMAIN, MINIMIZED AVERAGE STRUCTURE |
Matching Structure (courtesy of the PDB): |
|
| Term | Confidence | Notes |
| binding | 0.0980674097362609 | bayes_pls_golite062009 |
|
Region A: Residues: [148-270] |
1 11 21 31 41 51
| | | | | |
1 KHHKYSLTIL LWLVTFICSP CGVEGDHNPY VCLECSLMVH RDCIEDLPRV ISINRHDHRI 60
61 SHTFHLGERE GGWECGVCQT MINWVYGAYK CSRCPSYVVH SGCATKKEVW DGLDLEDVPD 120
121 EEE
|
| Detection Method: | |
| Confidence: | 2.19 |
| Match: | 1v5nA |
| Description: | Solution Structure of DC1 Domain of PDI-like Hypothetical Protein from Arabidopsis thaliana |
Matching Structure (courtesy of the PDB): |
|
| Term | Confidence | Notes |
| binding | 0.0196698141692984 | bayes_pls_golite062009 |
|
Region A: Residues: [271-488] |
1 11 21 31 41 51
| | | | | |
1 EIKDPFKVIN ENGDIVHISH EEHVLRLDEN YVTDDANMRC QCCILAINGD PCYRCVECDF 60
61 ILHDACANLP RKKQHLLHKH KLTLSSTCID GETYCQCKVY DVYTDGFIYK CHHEDCESKY 120
121 INYDARCSSV LEPFHHDLHQ HPLYFTLQSS RRCQACDREI HDDPLNCTVC DDYSLFMRCA 180
181 TLPRKVKHRC DDHFLSLCQG VGNAIGDLWC DICETKTN
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.