| Protein: | VID27_YEAST |
| Organism: | Saccharomyces cerevisiae S288c |
| Length: | 782 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for VID27_YEAST.
| Description | E-value | Query Range |
Subject Range |
|
|
0.0 | [1..782] | [1..782] |
|
|
0.0 | [12..768] | [16..780] |
|
|
0.0 | [12..768] | [18..782] |
|
|
0.0 | [243..768] | [112..572] |
|
|
0.0 | [265..768] | [138..646] |
|
|
0.0 | [312..766] | [16..492] |
|
|
9.0E-73 | [367..592] | [1..227] |
|
|
1.0E-4 | [458..622] | [40..192] |
|
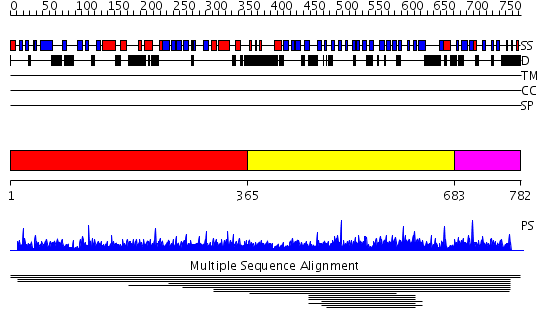
Region A: Residues: [1-364] |
1 11 21 31 41 51
| | | | | |
1 MNILKKFMES GNKPELITIP SGQFNLLRSK NSPKAALECI YNNATLSVRK IGKFDYELAV 60
61 YRVEDDSEGG TGDEAENFED DTISVLSTQS KKKEEEWSVE ISDKIMFHKT WDKQGNVALV 120
121 WENLRGDEQD EKVQFVVAAD VSFSDVEQFI QTVYRCQFEV RNKKSSLTAS ADDLKEIEHR 180
181 STRLFVQDDD DELDSSSDDF QDAKDTSFEH EKESEILERT PSPLKKVPEG EYCCLVMSSL 240
241 YMYDPIQEKF ILQEPVVKVA IIDTGKYEFW LAIEGKDNRL GTQVAPNINP TFELATDAFL 300
301 FNYTLQNITL SYMLKFKDLD KCIQFRFAWV KCLWMTLNKE TWTDVPEKEK DYILDSSSVP 360
361 LEKQ
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [365-682] |
1 11 21 31 41 51
| | | | | |
1 FDDILHIDDR SNEERDKESS ESENDSEDED DENDHSKRII SSEAFEEPRR ATSKGNSSLT 60
61 VAFRNNRSYV TRDNRIGVFK TDDEDDSLEF VAAIKNISNL GGKSIDPHKP MLYMEDRNLI 120
121 LTDGENENKL YKMDIERGKV IEEWSTGDKN VVQYGPTKKF DQMTPEQTIV GVSQKGVFKI 180
181 DPRINGKNKI AVDESKDYVG KYNFSSIGTT ESGYIAIGSE KGDIKLYDRL GIRAKTAIPS 240
241 LGQAIKFITT SADGKWLLAT CESTLLLMDL KIKDGKNAGN IGFLKSFPAS ENVKTYVLKI 300
301 RPEHSASILT YTKKPIRF
|
| Detection Method: | |
| Confidence: | 14.68 |
| Match: | 1a0rB |
| Description: | beta1-subunit of the signal-transducing G protein heterotrimer |
Matching Structure (courtesy of the PDB): |
|
| Term | Confidence | Notes |
| binding | 0.365473879287901 | bayes_pls_golite062009 |
|
Region A: Residues: [683-782] |
1 11 21 31 41 51
| | | | | |
1 TKAYFNTGIG QQEQTIVTST GPYAISWSLK GILNQDGSNN YPYRIRRYNA DVVADNFEFG 60
61 SDKKVIVALK DDVSLSKVKS FKQPSKGVLM PSASLQDFYG
|
| Detection Method: | |
| Confidence: | 10.5 |
| Match: | 1jjuB |
| Description: | Quinohemoprotein amine dehydrogenase B chain |
Matching Structure (courtesy of the PDB): |
|