| Protein: | KN12A_ARATH |
| Organism: | Arabidopsis thaliana |
| Length: | 1292 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for KN12A_ARATH.
| Description | E-value | Query Range |
Subject Range |
|
|
327.0 | [0..3] | [1292..2] |
|
|
321.0 | [0..169] | [1292..1764] |
|
|
318.0 | [0..86] | [444..2] |
|
|
314.0 | [0..91] | [827..11] |
|
|
313.0 | [0..82] | [460..5] |
|
|
308.0 | [0..86] | [1037..3] |
|
|
306.0 | [0..82] | [461..5] |
|
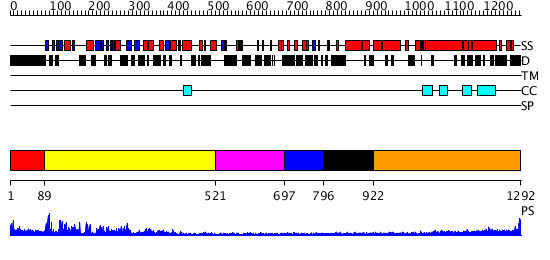
Region A: Residues: [1-88] |
1 11 21 31 41 51
| | | | | |
1 MKKHFTLPRN AILRDGGEPH SPNPSISKSK PPRKLRSAKE NAPPLDRNTS TPDHRSMRMK 60
61 NPLPPRPPPS NPLKRKLSAE TATESGFS
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [89-520] |
1 11 21 31 41 51
| | | | | |
1 DSGVKVIVRM KPLNKGEEGD MIVEKMSKDS LTVSGQTFTF DSIANPESTQ EQMFQLVGAP 60
61 LVENCLSGFN SSVFAYGQTG SGKTYTMWGP ANGLLEEHLC GDQRGLTPRV FERLFARIKE 120
121 EQVKHAERQL NYQCRCSLLE IYNEQITDLL DPSQKNLMIR EDVKSGVYVE NLTEEYVKNL 180
181 TDVSQLLIKG LGNRRTGATS VNTESSRSHC VFTCVVESRC KNVADGLSSF KTSRINLVDL 240
241 AGSERQKSTG AAGERLKEAG NINRSLSQLG NLINILAEIS QTGKPRHIPY RDSRLTFLLQ 300
301 ESLGGNAKLA MVCAVSPSQS CRSETFSTLR FAQRAKAIQN KAVVNEVMQD DVNFLRGVIH 360
361 QLRDELQRMK NDGNNPTNPN VAYSTAWNAR RSLNLLRSFG LGHPRSLPHE DNDGDIEMEI 420
421 DEAAVERLCV QV
|
| Detection Method: | |
| Confidence: | 86.30103 |
| Match: | 1gojA |
| Description: | Kinesin |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [521-696] |
1 11 21 31 41 51
| | | | | |
1 GLQSSLASEG INHDMNRVKS IHSSDGQSIE KRLPEDSDVA MEDACCHTEN HEPETVDNMR 60
61 TETETGIREN QIKTHSQTLD HESSFQPLSV KDALCSSLNK SEDVSSCPDL VPQDVTSANV 120
121 LIADGVDDPE HLVNSASPSL CIDPVGATPV LKSPTLSVSP TIRNSRKSLK TSELST
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [697-795] |
1 11 21 31 41 51
| | | | | |
1 ASQKDSEGEN LVTEAADPSP ATSKKMNNCS SALSTQKSKV FPVRTERLAS SLHKGIKLLE 60
61 SYCQSTAQRR STYRFSFKAP DSEPSTSISK ADAGVQTIP
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [796-921] |
1 11 21 31 41 51
| | | | | |
1 GADAISEENT KEFLCCKCKC REQFDAQQMG DMPNLQLVPV DNSEVAEKSK NQVPKAVEKV 60
61 LAGSIRREMA LEEFCTKQAS EITQLNRLVQ QYKHERECNA IIGQTREDKI IRLESLMDGV 120
121 LSKEDF
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
| MCM Score |
SCOP Match |
SCOP Description | ||
| View | Download | 0.902 | a.24.9 | alpha-catenin/vinculin |
|
Region A: Residues: [922-1292] |
1 11 21 31 41 51
| | | | | |
1 LDEEFASLLH EHKLLKDMYQ NHPEVLKTKI ELERTQEEVE NFKNFYGDMG EREVLLEEIQ 60
61 DLKLQLQCYI DPSLKSALKT CTLLKLSYQA PPVNAIPESQ DESLEKTLEQ ERLCWTEAET 120
121 KWISLSEELR TELEASKALI NKQKHELEIE KRCGEELKEA MQMAMEGHAR MLEQYADLEE 180
181 KHMQLLARHR RIQDGIDDVK KAAARAGVRG AESRFINALA AEISALKVEK EKERQYLRDE 240
241 NKSLQTQLRD TAEAIQAAGE LLVRLKEAEE GLTVAQKRAM DAEYEAAEAY RQIDKLKKKH 300
301 ENEINTLNQL VPQSHIHNEC STKCDQAVEP SVNASSEQQW RDEFEPLYKK ETEFSNLAEP 360
361 SWFSGYDRCN I
|
| Detection Method: | |
| Confidence: | 9.522879 |
| Match: | 1i84S |
| Description: | Heavy meromyosin subfragment |
Matching Structure (courtesy of the PDB): |
|