| Protein: | CG32000-PA, CG32... |
| Organism: | Drosophila melanogaster |
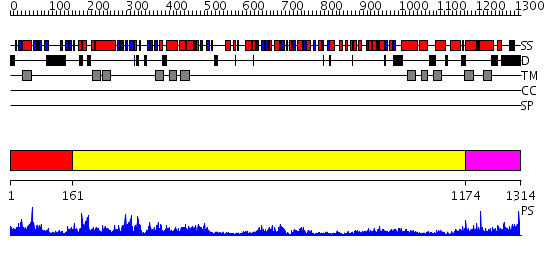
| Length: | 1314 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for CG32000-PA, CG32....
| Description | E-value | Query Range |
Subject Range |
|
|
785.0 | [0..9] | [1314..83] |
|
Region A: Residues: [1-160] |
1 11 21 31 41 51
| | | | | |
1 MTPHNDSKDS KPCIGLLNPD QEDQMKVCGY RRSLMRTGFC WACIFLTGGL LRLVLHWWRH 60
61 LYLYATCSQC SLEEAEQVLV TEDYQGKHKM YHVKQIQVLT SSNLKTLLEK EQQSIERTHI 120
121 ECDHVENVLQ LSVHFTSAQF KKCSSIRIFR CKQLVYAWNN
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [161-1173] |
1 11 21 31 41 51
| | | | | |
1 NTNRFQRING LDLNIPCSYY HQQRGLPVHE QISRRIVFGD NEITVPLRDF KTLLFLEVLN 60
61 PFYVFQLFSV ILWFTYDYYY YACVILLMSV FGITVSVLQT KKNQDVLQKT VYNTGNAWVV 120
121 DHKGLSKELP TRAIVPGDII EIPSSGCTLH CDAILISGNC ILDESMLTGE SVPVTKTPLP 180
181 SKRDMIFDKT EHARHTLFCG TKVIQTRYIG SKKVLAFVIN TGNITAKGEL IRSILYPPPV 240
241 DYKFEQDSYK FIQFLAIIAC VGFIYTLVTK ILRGTDPVKI AVESLDLITI VVPPALPAAM 300
301 TVGRFYAQKR LKTSEIFCIS PRSINVAGSI NCCCFDKTGT LTEDGLDMWG VVPKSSTNQF 360
361 QIPLKSVDRL PFDHFLFGMV TCHSITILNG RMMGDPLDLK MFESTGWELE DSNNIPDTEK 420
421 YGILYPTILR QPRGGLSGMA ETESGSKNEI KRQSSVDDLL ATVGISPSQK NFDHGIVREF 480
481 PFTSALQRMS VVTRCLSDQV FNVYCKGSPE MLKKLCKPQS LPDNYSQQLS EFAKKGYRII 540
541 AIAFKALSHK MNYTKVQRLS REEVENNMEF LGFVILENRL KPDTTKVINA LNAAKIRTIM 600
601 ITGDNILTAI SVARDCGIVS PSQSVITVHA DPIGDSANIQ TNTGTECNFD NSSDKHYKLH 660
661 YTLDLGSKTS RAYLFKSCFN SNLFDPETPE FTAQVGKTIF HMESTNSLVN ESTSSYAESG 720
721 LPTSDSLASV KTIDTWTHND AELGIKHTPD ESWRRQECIF AMDGKTWQIV KDYFPEEMEI 780
781 LLTRGSIYAR MSPDQKQALV IELQNLDYCV AMCGDGANDC GALKVAHAGI SLSETEASIA 840
841 SPFTSRNPTI SAVLKVIKEG RAALVTSFGI FKYMAAYSLV QFISVMILYS IDSNLTDKQY 900
901 LYVDLGLISI FAFFFGKTES FDGMLVEQVP LSSLISSTPL ASLLLHLTVV TAFQVTCWVH 960
961 LHQQPWFKAF EPADEDHLGC FENYTMFCIS SFQYIILAFV FSKGAPYRKP LWS
|
| Detection Method: | |
| Confidence: | 152.0 |
| Match: | 1iwoA |
| Description: | Calcium ATPase, transduction domain A; Calcium ATPase, catalytic domain P; Calcium ATPase; Calcium ATPase, transmembrane domain M |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [1174-1314] |
1 11 21 31 41 51
| | | | | |
1 NWPLCLAFIV NLCIIVYLVL YPSDWVASFF QLIVPPTMRF RYVMLAYGAA SFICHIFVES 60
61 FLVEYLVFKK YQVKREKNWV TSKQKYMRLE HDISNIKNWP PITEVYEPNN LIDCETEQPT 120
121 YVSLHAEQNH DTQLGKFPGF C
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.