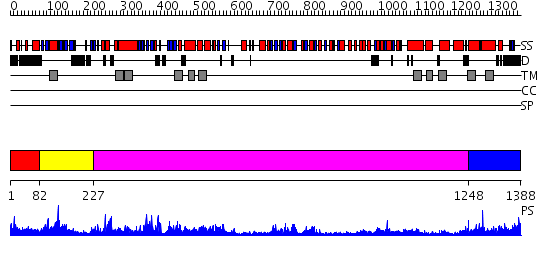
| Protein: | CG32000-PG |
| Organism: | Drosophila melanogaster |
| Length: | 1388 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for CG32000-PG.
| Description | E-value | Query Range |
Subject Range |
|
|
832.0 | [0..68] | [1388..1] |
|
Region A: Residues: [1-81] |
1 11 21 31 41 51
| | | | | |
1 MHYVATESVQ PKKSDKVPSN KIKKVENNNT LVNGCSKTSA RSVPLLKYNR PDQDGDSEEN 60
61 ITSVLEPNVD EIYSKDSERL V
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [82-226] |
1 11 21 31 41 51
| | | | | |
1 DDSKPCIGLL NPDQEDQMKV CGYRRSLMRT GFCWACIFLT GGLLRLVLHW WRHLYLYATC 60
61 SQCSLEEAEQ VLVTEDYQGK HKMYHVKQIQ VLTSSNLKTL LEKEQQSIER THIECDHVEN 120
121 VLQLSVHFTS AQFKKCSSIR IFRCK
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [227-1247] |
1 11 21 31 41 51
| | | | | |
1 QLVYAWNNNT NRFQRINGLD LNIPCSYYHQ QRGLPVHEQI SRRIVFGDNE ITVPLRDFKT 60
61 LLFLEVLNPF YVFQLFSVIL WFTYDYYYYA CVILLMSVFG ITVSVLQTKK NQDVLQKTVY 120
121 NTGNAWVVDH KGLSKELPTR AIVPGDIIEI PSSGCTLHCD AILISGNCIL DESMLTGESV 180
181 PVTKTPLPSK RDMIFDKTEH ARHTLFCGTK VIQTRYIGSK KVLAFVINTG NITAKGELIR 240
241 SILYPPPVDY KFEQDSYKFI QFLAIIACVG FIYTLVTKIL RGTDPVKIAV ESLDLITIVV 300
301 PPALPAAMTV GRFYAQKRLK TSEIFCISPR SINVAGSINC CCFDKTGTLT EDGLDMWGVV 360
361 PKSSTNQFQI PLKSVDRLPF DHFLFGMVTC HSITILNGRM MGDPLDLKMF ESTGWELEDS 420
421 NNIPDTEKYG ILYPTILRQP RGGLSGMAET ESGSKNEIKR QSSVDDLLAT VGISPSQKNF 480
481 DHGIVREFPF TSALQRMSVV TRCLSDQVFN VYCKGSPEML KKLCKPQSLP DNYSQQLSEF 540
541 AKKGYRIIAI AFKALSHKMN YTKVQRLSRE EVENNMEFLG FVILENRLKP DTTKVINALN 600
601 AAKIRTIMIT GDNILTAISV ARDCGIVSPS QSVITVHADP IGDSANIQTN TGTECNFDNS 660
661 SDKHYKLHYT LDLGSKTSRA YLFKSCFNSN LFDPETPEFT AQVGKTIFHM ESTNSLVNES 720
721 TSSYAESGLP TSDSLASVKT IDTWTHNDAE LGIKHTPDES WRRQECIFAM DGKTWQIVKD 780
781 YFPEEMEILL TRGSIYARMS PDQKQALVIE LQNLDYCVAM CGDGANDCGA LKVAHAGISL 840
841 SETEASIASP FTSRNPTISA VLKVIKEGRA ALVTSFGIFK YMAAYSLVQF ISVMILYSID 900
901 SNLTDKQYLY VDLGLISIFA FFFGKTESFD GMLVEQVPLS SLISSTPLAS LLLHLTVVTA 960
961 FQVTCWVHLH QQPWFKAFEP ADEDHLGCFE NYTMFCISSF QYIILAFVFS KGAPYRKPLW1020
1021 S
|
| Detection Method: | |
| Confidence: | 165.0 |
| Match: | 1iwoA |
| Description: | Calcium ATPase, transduction domain A; Calcium ATPase, catalytic domain P; Calcium ATPase; Calcium ATPase, transmembrane domain M |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [1248-1388] |
1 11 21 31 41 51
| | | | | |
1 NWPLCLAFIV NLCIIVYLVL YPSDWVASFF QLIVPPTMRF RYVMLAYGAA SFICHIFVES 60
61 FLVEYLVFKK YQVKREKNWV TSKQKYMRLE HDISNIKNWP PITEVYEPNN LIDCETEQPT 120
121 YVSLHAEQNH DTQLGKFPGF C
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.