| Protein: | gi|45445605, gi|... |
| Organism: | Drosophila melanogaster |
| Length: | 807 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for gi|45445605, gi|....
| Description | E-value | Query Range |
Subject Range |
|
|
500.0 | [0..1] | [807..1] |
|
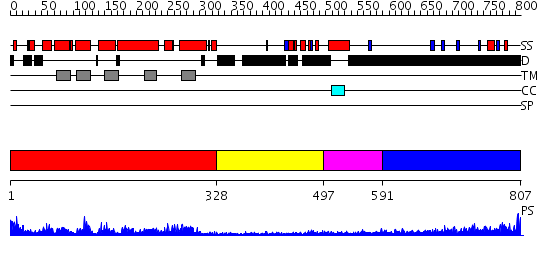
Region A: Residues: [1-327] |
1 11 21 31 41 51
| | | | | |
1 MDPDNDIYAF YDIRGYKGKC RPGRPNSERI LQPRMSLLGK PLNYNRGTRR DVRYRRLQSR 60
61 LYNFLERPRG LHAIFYHVMV FLMVFTCLAL SVFSTIKEYE EDAVYILFRM EILVVIWFTM 120
121 EFGARLWSSG CRSRYQGCLG RLKFVKRPFC IIDIVTILAS IVVLGMGTSG QVFATSALRG 180
181 LRFFQILRMV RMDRRGGTWK LLGSVVYAHR QELITTMYIG FLGLIFASFL VYMWEKDVND 240
241 KFSNFAQALW WGITLCTVGY GDMVPITWQG KLIASCCALL GISFFALPAG ILGSGFALKV 300
301 QQQQRQKHMI RRRQPAATLI QAVWRCY
|
| Detection Method: | |
| Confidence: | 69.39794 |
| Match: | 2a79B |
| Description: | Mammalian Shaker Kv1.2 potassium channel- beta subunit complex |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [328-496] |
1 11 21 31 41 51
| | | | | |
1 AADEHSVSVA TWNIHRVALP SPPASRASSS FKHNTSFVAR LPTIRRHKSQ TIQTPGGGDG 60
61 GGVSKPPGSS RASTRYTRTI RDINASVENL DEEDEPRCTQ LTNRHKTAIR FIRKLKYFVA 120
121 RRKFKEALKP YDVKDVMEQY AAGHVDLLGR VKMLHLRLDQ ILGKQGSKA
|
| Detection Method: | |
| Confidence: | 5.154902 |
| Match: | 2qksA |
| Description: | No description for 2qksA was found. |
|
Region A: Residues: [497-590] |
1 11 21 31 41 51
| | | | | |
1 KDVYASKISL ASRVVKVERQ VADIEEKLDI LIKAYMEDRD RFLALPLPAK PKIHSISPSH 60
61 KPLHHAHNLA MIDVWKRTAA LSVHPEQVTT TPLL
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [591-807] |
1 11 21 31 41 51
| | | | | |
1 NPSAPDSSEL RSLTATQTPT TTTDAIATQT PMPPHVQHTA TNTKSSVLNS YQLGSEKQQH 60
61 NDVFMTELEN RTKKRVTLSL HRSTSEPYSK QEQRITIPDE GADSLDSSAK PTPPDSSIIL 120
121 IDEYEDFEEE DLNCEGEMDH FPTWEIDSDI GVEVDVDADA DGDCDESTED TALLQCATRT 180
181 AIVITPISPV SSAHNLQQLN DQTTTLNKSN LLPPDSG
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.