| Protein: | NUP133 |
| Organism: | Homo sapiens |
| Length: | 1156 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for NUP133.
| Description | E-value | Query Range |
Subject Range |
|
|
0.0 | [1..1156] | [1..1156] |
|
|
0.0 | [1..1156] | [1..1156] |
|
|
0.0 | [1..1156] | [1..1155] |
|
|
0.0 | [1..1156] | [1..1156] |
|
|
0.0 | [1..1156] | [1..1154] |
|
|
0.0 | [7..1156] | [3..1136] |
|
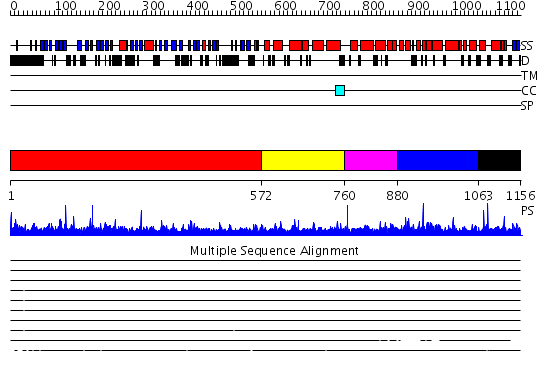
Region A: Residues: [1-571] |
1 11 21 31 41 51
| | | | | |
1 MFPAAPSPRT PGTGSRRGPL AGLGPGSTPR TASRKGLPLG SAVSSPVLFS PVGRRSSLSS 60
61 RGTPTRMFPH HSITESVNYD VKTFGSSLPV KVMEALTLAE VDDQLTINID EGGWACLVCK 120
121 EKLIIWKIAL SPITKLSVCK ELQLPPSDFH WSADLVALSY SSPSGEAHST QAVAVMVATR 180
181 EGSIRYWPSL AGEDTYTEAF VDSGGDKTYS FLTAVQGGSF ILSSSGSQLI RLIPESSGKI 240
241 HQHILPQGQG MLSGIGRKVS SLFGILSPSS DLTLSSVLWD RERSSFYSLT SSNISKWELD 300
301 DSSEKHAYSW DINRALKENI TDAIWGSESN YEAIKEGVNI RYLDLKQNCD GLVILAAAWH 360
361 SADNPCLIYY SLITIEDNGC QMSDAVTVEV TQYNPPFQSE DLILCQLTVP NFSNQTAYLY 420
421 NESAVYVCST GTGKFSLPQE KIVFNAQGDS VLGAGACGGV PIIFSRNSGL VSITSRENVS 480
481 ILAEDLEGSL ASSVAGPNSE SMIFETTTKN ETIAQEDKIK LLKAAFLQYC RKDLGHAQMV 540
541 VDELFSSHSD LDSDSELDRA VTQISVDLMD D
|
| Detection Method: | |
| Confidence: | 165.0 |
| Match: | 1xksA |
| Description: | The crystal structure of the N-terminal domain of Nup133 reveals a beta-propeller fold common to several nucleoporins |
Matching Structure (courtesy of the PDB): |
|
| Term | Confidence | Notes |
| nucleocytoplasmic transporter activity | 7.36869426424539 | bayes_pls_golite062009 |
| transporter activity | 2.55928656852707 | bayes_pls_golite062009 |
| binding | 1.5683135853172 | bayes_pls_golite062009 |
| protein binding | 0.571876690720183 | bayes_pls_golite062009 |
|
Region A: Residues: [572-759] |
1 11 21 31 41 51
| | | | | |
1 YPASDPRWAE SVPEEAPGFS NTSLIILHQL EDKMKAHSFL MDFIHQVGLF GRLGSFPVRG 60
61 TPMATRLLLC EHAEKLSAAI VLKNHHSRLS DLVNTAILIA LNKREYEIPS NLTPADVFFR 120
121 EVSQVDTICE CLLEHEEQVL RDAPMDSIEW AEVVINVNNI LKDMLQAASH YRQNRNSLYR 180
181 REESLEKE
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [760-879] |
1 11 21 31 41 51
| | | | | |
1 PEYVPWTATS GPGGIRTVII RQHEIVLKVA YPQADSNLRN IVTEQLVALI DCFLDGYVSQ 60
61 LKSVDKSSNR ERYDNLEMEY LQKRSDLLSP LLSLGQYLWA ASLAEKYCDF DILVQMCEQT 120
121
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [880-1062] |
1 11 21 31 41 51
| | | | | |
1 DNQSRLQRYM TQFADQNFSD FLFRWYLEKG KRGKLLSQPI SQHGQLANFL QAHEHLSWLH 60
61 EINSQELEKA HATLLGLANM ETRYFAKKKT LLGLSKLAAL ASDFSEDMLQ EKIEEMAEQE 120
121 RFLLHQETLP EQLLAEKQLN LSAMPVLTAP QLIGLYICEE NRRANEYDFK KALDLLEYID 180
181 EEE
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [1063-1156] |
1 11 21 31 41 51
| | | | | |
1 DININDLKLE ILCKALQRDN WSSSDGKDDP IEVSKDSIFV KILQKLLKDG IQLSEYLPEV 60
61 KDLLQADQLG SLKSNPYFEF VLKANYEYYV QGQI
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.