| Protein: | SPBC887.12 |
| Organism: | Schizosaccharomyces pombe |
| Length: | 1258 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for SPBC887.12.
| Description | E-value | Query Range |
Subject Range |
|
|
876.0 | [0..21] | [1257..93] |
|
|
861.0 | [0..21] | [1257..95] |
|
|
857.0 | [0..29] | [1257..99] |
|
|
855.0 | [0..12] | [1257..109] |
|
|
854.0 | [0..27] | [1257..101] |
|
|
847.0 | [0..21] | [1257..94] |
|
|
840.0 | [0..55] | [1257..137] |
|
|
830.0 | [0..4] | [1258..47] |
|
|
829.0 | [0..33] | [1257..108] |
|
|
825.0 | [0..41] | [1257..62] |
|
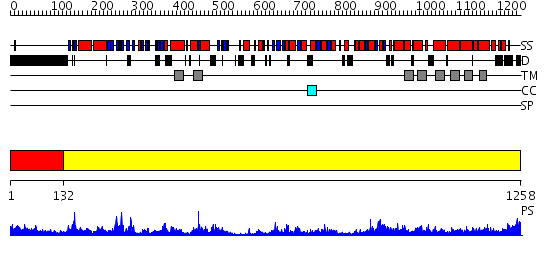
Region A: Residues: [1-131] |
1 11 21 31 41 51
| | | | | |
1 MARDVDNKQN AKRISRDEDE DEFAGESMVG RTLDNPFLGE DEFEDIFGSE SQYISSSGQN 60
61 STNPFLADTR IENSPLGSES KANQLNKQGT NVNHIEIPLR DFNDPTQPES FLPPPKNTFT 120
121 SRIKKIKNLF K
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [132-1258] |
1 11 21 31 41 51
| | | | | |
1 KEKKQVKPED LGPRQIILND YSANHFLHNA VSTCKYSAFT FLPKFLKEQF SKYANLFFLF 60
61 TAVVQQIPGI TPVNRYTTIG PMLIVLSVSG IKEIMEDIKR KKQDQELNES PCYVLQGTGF 120
121 VEKQWKDVVV GDIVKIVSET FFPADLVLLS SSEPEGLCYI ETANLDGETN LKIKQALPET 180
181 AGLLKPVELG QLSGEVKSEQ PNNNLYTFDA TLKLLPSDRE LPLSPDQLLL RGAQLRNTPW 240
241 VYGIVVFTGH ESKLMKNTTE TPIKRTSVEK QVNSQILFLL CIFVFLCFAS SLGALIHRSV 300
301 YGSALSYVKY TSNRAGMFFK GLLTFWILYS NLVPISLFVT FELVRYIQAQ LISSDLDMYN 360
361 EETDTPAACR TSSLVEELGQ VGYIFSDKTG TLTRNQMEFR QCTIAGVAYA DVIPEDRQFT 420
421 SEDLDSDMYI YDFDTLKENL KHSENASLIH QFLLVLSICH TVIPEYDEST NSIKYQASSP 480
481 DEGALVKGAA SIGYKFLARK PHLVTVSIFG KDESYELLHI CEFNSTRKRM SIVFRCPDGK 540
541 IRLYVKGADT VIMERLASDN PYLQTTIHHL EDYATVGLRT LCIAMREVPE DEYQRWSTVF 600
601 ETAASSLVDR AQKLMDAAEE IEKDLILLGA TAIEDRLQDG VPDTISTLQT AGIKIWVLTG 660
661 DRQETAINIG MSCKLIDEDM GLVIVNEETK EATAESVMAK LSSIYRNEAT TGNVESMALV 720
721 IDGVSLTYAL DFSLERRFFE LASLCRAVIC CRVSPLQKAL IVKMVKRNTG EVLLAIGDGA 780
781 NDVPMIQAAH VGVGISGMEG LQAVRSSDFS ISQFCYLKKL LLVHGSWCYQ RLSKLILYSF 840
841 YKNIALYMTQ FWYAFCNAFS GQVIFESWSI SLYNVLFTVL PPVVIGIFDQ FVSAGQLFQY 900
901 PQLYQLGQRS EFFNLKRFWS WITNGFYHSL LLFLCSIAVF YYDGPNKDGL ASGHWVWGTT 960
961 LYAAILATVL GKAALISNHW TQYTVIATLG SFLLWIVFMP IYAVAAPAIG FSKEYYGIIP1020
1021 HLYGNLKFWA SLLVLPTIAL MRDFVWKYSS RMYYPEEYHY VQEIQKYNVT DYRPRIVGFH1080
1081 KAIRKIRQMQ RMRKQRGYAF SQGEEDQSRI LDAYDTTHTR GAYGEMR
|
| Detection Method: | |
| Confidence: | 163.0 |
| Match: | 1iwoA |
| Description: | Calcium ATPase, transduction domain A; Calcium ATPase, catalytic domain P; Calcium ATPase; Calcium ATPase, transmembrane domain M |
Matching Structure (courtesy of the PDB): |
|