| Protein: | SPAC631.02 |
| Organism: | Schizosaccharomyces pombe |
| Length: | 727 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for SPAC631.02.
| Description | E-value | Query Range |
Subject Range |
|
|
304.0 | [0..88] | [703..1] |
|
|
303.0 | [0..213] | [713..11] |
|
|
300.0 | [0..216] | [692..13] |
|
|
296.0 | [0..146] | [700..2] |
|
|
292.0 | [0..363] | [713..11] |
|
|
284.0 | [0..162] | [700..1063] |
|
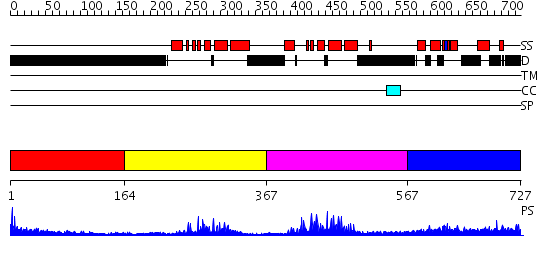
Region A: Residues: [1-163] |
1 11 21 31 41 51
| | | | | |
1 MTPVQVPNLD VSVTSSLITT APVTGNAATI STFTPGSPEP SMNGEEKESS YFPISENDDG 60
61 TLDLFGDSEL EKEQKGDNQE TDYSSQYLHP TPPYTNFDDE SPSSPTHPSV SNITVDGDSK 120
121 KHSLQLQEEE KSSESLDSHT HPPKRVRNED DSLTFSKTSP VSP
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [164-366] |
1 11 21 31 41 51
| | | | | |
1 SSLKDGASNT VTNDASNKIK SEASESASPS ALQALDSTAA GSSKEHSSPH DETVKKEEND 60
61 KDQYPPMTKE QHKYIHAMLR QLRRGRDSIP FRAPVDPVKQ NIPDYPTIIK NPIDLGTMQK 120
121 KFSSGVYSSA QHFIDDMNLM FSNCFLYNGT ESPVGVMGKN LQATFERQLK QLPSAYVTSY 180
181 SRPGRRPRSM TAPKGGARTR RQA
|
| Detection Method: | |
| Confidence: | 13.522879 |
| Match: | 1z4rA |
| Description: | Human GCN5 Acetyltransferase |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [367-566] |
1 11 21 31 41 51
| | | | | |
1 AMYSNSSSGI RETMYDLKPH RRKDAAEMKF CQSVLKELLK KQHEAYAYPF YKPVNPTACG 60
61 CPDYFKVIKH PMDLGTMQNK LNHNEYASMK AFEADMVLMF KNCYKFNSAG TPVHLMGKKL 120
121 ESIFQKLWAN KPDFDSETYM GMSSVNTDYY YGDNEVFDSG DEFLEDDGEE FEAVNRQIHK 180
181 LQSTLQAMKS RARSSSVSRR
|
| Detection Method: | |
| Confidence: | 36.69897 |
| Match: | 2dvvA |
| Description: | No description for 2dvvA was found. |
|
Region A: Residues: [567-727] |
1 11 21 31 41 51
| | | | | |
1 SRSRSLSVDI YPPITYEMQN ELAEQCNYLS ADQLSHVAEI LRAALPHLRN TDEIEIDVSA 60
61 MPPDVFYKVY YYVCKGDEIG AEALATASHT HQEKKKGRAL SETEQAEKIR QLRAQLDRFS 120
121 GIAQNKNTVT GNIAAYNTKS LGSDDSSSED DGESSESSDS A
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.