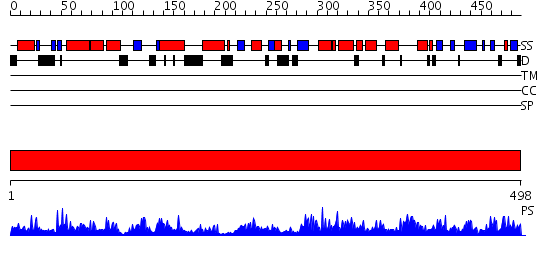
| Protein: | gsa1 |
| Organism: | Schizosaccharomyces pombe |
| Length: | 498 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for gsa1.
| Description | E-value | Query Range |
Subject Range |
|
|
756.0 | [0..1] | [498..1] |
|
|
708.0 | [0..2] | [497..87] |
|
|
704.0 | [0..2] | [497..83] |
|
|
699.0 | [0..6] | [497..85] |
|
|
698.0 | [0..1] | [497..60] |
|
|
685.0 | [0..2] | [496..70] |
|
Region A: Residues: [1-498] |
1 11 21 31 41 51
| | | | | |
1 MEIEKYTPEQ IEELGKGARD FAFAHGVVFT ELSVSKEGRN IATQIPITLF PSVIPHGAFV 60
61 EAVSVQKAYN KLYAKIANDY EFLRLHLQSI TKYDEFMNKL WNLYQKHREA VAHLKENQFQ 120
121 PLSLGVFRSD YMVHQDDSFI GCKQVEFNTI SVSFGGVSKA VSNLHAYCSQ SGLYRKPLTT 180
181 NYLTVNTSVS GICTGISNAV DAYRDYVKNI TSKMNIASDN TKPIVLFVVK GGERNITDQR 240
241 TLEYELLNRF HVISKRIDIA ELNSLIHDKS SNKLYMKTSF TTYEVAVVYY RVGYALDDYP 300
301 SQEAWDMRLT IENTLAIKCP SISTHLAGSK KIQQVLAESN ALERFLEGDE LQAVRSTFAD 360
361 MYPLDDTPRG KEGIKLAFEK PEDFVLKPQR EGGGNNTYGK DIPGLLSKMP QEEWDSYILM 420
421 RYINAVPSQN YILKGERPEK FDVVDEIGIL GTIVWNIKTD EVVQNGQSGF ICRTKPKKTN 480
481 EGGVATGYAS LSSIELSE
|
| Detection Method: | |
| Confidence: | 165.0 |
| Match: | 1m0tA |
| Description: | Eukaryotic glutathione synthetase, substrate-binding domain; Eukaryotic glutathione synthetase ATP-binding domain |
Matching Structure (courtesy of the PDB): |
|