| Protein: | SPAC29A4.19c |
| Organism: | Schizosaccharomyces pombe |
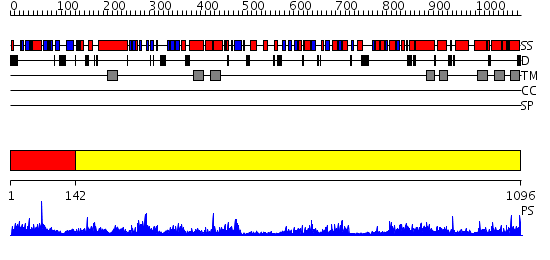
| Length: | 1096 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for SPAC29A4.19c.
| Description | E-value | Query Range |
Subject Range |
|
|
742.0 | [0..135] | [1096..153] |
|
|
733.0 | [0..127] | [1096..21] |
|
|
731.0 | [0..126] | [1096..20] |
|
|
730.0 | [0..126] | [1096..20] |
|
|
730.0 | [0..126] | [1096..20] |
|
|
730.0 | [0..106] | [1096..1] |
|
|
729.0 | [0..120] | [1096..3] |
|
|
729.0 | [0..126] | [1096..15] |
|
|
728.0 | [0..135] | [1096..31] |
|
Region A: Residues: [1-141] |
1 11 21 31 41 51
| | | | | |
1 MDSIELKQLV PENDSEPGTP RQLLFQHYDI SNEETIGIKP FKSIPAKVYI LRVTEILTLG 60
61 LLHLILTWLP EFRLKWIEAP CSNEDVEFVA ISDPSGTSSI EKVSSICLKN DIQTSSFVLP 120
121 SGKTRYFEYK KLRFYLEPLN L
|
| Detection Method: |
Shown below are all of our structure predictions for this domain.
Click here to view only most confident match.
| MCM Score |
GO Score |
GO Term |
SCOP Match |
SCOP Description | ||
| View | Download | 0.207 | 0.043 | ATPase activity, coupled to transmembrane movement of ions, phosphorylative mechanism | d.93.1 | SH2 domain |
| View | Download | 0.207 | N/A | N/A | d.93.1 | SH2 domain |
|
Region A: Residues: [142-1096] |
1 11 21 31 41 51
| | | | | |
1 QWVLMPLETS AYSLVTSTPA YIQNGLDTFT IAKLRQVYGS NSLVSTKKSI VTILLNEVLH 60
61 PFYLFQAVSV LIWLCDSFVF YSCCIVFISS YSIFLSVKES KESENRIHSI IGAPQPVTVI 120
121 RNQVKQTVLA DDLVIGDLLY FSNLDLKTCP VDGILFSSSC LLDESMVTGE SVPARKFPLE 180
181 DNSLDSWMIA SCNIFSPHLI HAGTKFLKID STPSTPCLIS VVRTGFRSNK GQLIRNLLYP 240
241 NLRPSQLYLD SMSFLKTMAI LSFVSIVFIA IYLNLYNASF GHVVLRSLDV LTILVPPALP 300
301 ATLSVGIANS IARLSRALIY TTSPESIHNA GCLSTFVFDK TGTLTENSVQ LSCVYVKSGS 360
361 NGLLKQVDAD SLSLDSTKLN AHAYRVATCS QSLELVGNEL VGDPLEVTLF TQFNGTFCAT 420
421 IRASNTPHPP LFSVSNSFDG PSQIFSIYKA LEFDPVLRRM SVICSTSTER SLMLFTKGAP 480
481 ESILAISSQQ SIPSNVQEVI HTLSSKGFRI IAFASKNLIT PLQELIHLSR STLESNVTFQ 540
541 GLFVLESPLR ESSKDVISSL LRSKMEVSIC SGDSLFTSVF VAKHCGALDS CNFIYTAELA 600
601 DSGDDCPQIH FEKIDLQTQN FQPIPDGFSL KDVILEKDSS LCMDGKLLQR LLTMLSFNEI 660
661 KILLSKLRVL ARMSPFDKAT YVELCQKYGC KVGFCGDGAN DCIALKQADV GVSLSDSEAC 720
721 AAASFVSKKK SIKDVFNVLL EGRCSLILSH RCFQYMVLCA IVQFSGVFFL YLKNYNFNDN 780
781 QFLFMDLLII FPLSAAMSYF DPAQNLTSNR PNSTLFGKGR VKDLGIQSVL IWLSHGLLTL 840
841 ILHELNWVEL PEWQLEKSNT KNVLVTSIFL LSSLQYLGIC IGINQSSEFL SPIWKKKTYV 900
901 CLCTTIGLCN IYLCFANENH IISRCLQITR LPTLYRFIIL FMGVISCCLT SILNM
|
| Detection Method: | |
| Confidence: | 164.0 |
| Match: | 1iwoA |
| Description: | Calcium ATPase, transduction domain A; Calcium ATPase, catalytic domain P; Calcium ATPase; Calcium ATPase, transmembrane domain M |
Matching Structure (courtesy of the PDB): |
|