| Protein: | cta4 |
| Organism: | Schizosaccharomyces pombe |
| Length: | 1211 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for cta4.
| Description | E-value | Query Range |
Subject Range |
|
|
776.0 | [0..1] | [1211..1] |
|
|
721.0 | [0..97] | [1091..9] |
|
|
721.0 | [0..97] | [1091..9] |
|
|
720.0 | [0..97] | [1091..9] |
|
|
720.0 | [0..97] | [1091..131] |
|
|
719.0 | [0..97] | [1091..9] |
|
|
718.0 | [0..97] | [1091..9] |
|
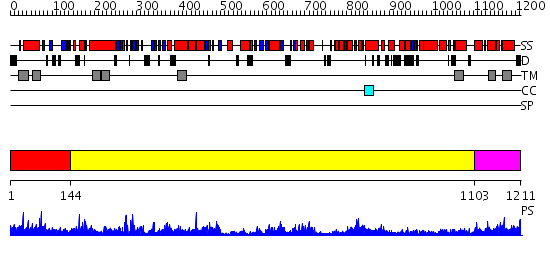
Region A: Residues: [1-143] |
1 11 21 31 41 51
| | | | | |
1 MGSKALITSP DISSGQLYIK LPTFFHLYVW PFALFVYPYI GYVYQNKLYS EEVRYLTYIA 60
61 VGTIHALFWL AGEWNTKVYC LMTCRKTDKV EQATHILVTP SKIGESSSVE PITKLVLPDS 120
121 QTIQYSFSFQ RKRFIYEPEK GCF
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [144-1102] |
1 11 21 31 41 51
| | | | | |
1 ANITFPMDEP STIGTLKKST GLTNIQSEIF LYRYGKNCFD IPIPTFGTLF KEHAVAPFFV 60
61 FQIFCCVLWC LDDYWYFSLF SMFMIIALEC SVVWQRQRTL TEFRTMSIKP YEIQVYRNKH 120
121 WFPISTEDLL PNDVVSVLHN KEDSGLPCDL LLLSGSCVVN EAMLSGESTP LVKESIELRP 180
181 EEAVIDVDEL DKNAVLFGGT RVLQVTQSPF CKLKTPDNGV PAIVLRTGFE TSQGSLVRTM 240
241 VFSSEKVTAN NRESLYFILF LLVFAIAASG YVWHVGSKTE RSRYKLMLDC VMIITSVVPS 300
301 ELPMELSMAV NASLGALSKY YIYCTEPFRI PLSGHLDICC FDKTGTLTEE HMVVQGIAGV 360
361 NRKDPYSLEK LSDASNDAIL AIATAHTLVL LEQEGETPKV VGDPMEKATV ENLGWSIEKK 420
421 NFVSAPEGSV FYKGKVQIIR NFQFSSALKR QSSVSNVRVS GGSFKTFVSV KGAPEVIATM 480
481 LREVPKDYEK IYKDYGRKGS RVLALGYKYF KNYIPENQVS DLSRESIESD LVFAGFLIFT 540
541 SPLKEDARQT VQMLNNSSHR CMMITGDNPL TAVYVAEQVG IVEKPTLVLD IKHENEKILE 600
601 WKSTDDTINL PMNPHKSLEA SLYEKYDLCI TGRALSQIIN PDVIMSIFTH AWVYARVSPS 660
661 QKEFMISTLK HNGYITLMCG DGTNDVGALK QAHVGVALLN ASEEDMLEMQ ERARNQKLMG 720
721 VYEKQIQLAK RFNLPTPPVP PALCHAFPPG PNNPHREKTQ EGLNKVLEDL ETKKASDVQL 780
781 TEAEKAAERR ANLANKMFDT LANASDDEAP KLKLGDASVA APFTSKLAVV SSITNIVRQG 840
841 RCTLVALVQM HKILALNCLI TAYSLSVLHL DGIKFGDTQY TISGMLMSVC FYCVSRARPL 900
901 ETLSKERPQA GIFNTYIIGS VLGQFAIHIV TLIYITRVVY LYEDPLEKVD LEETFKPSL
|
| Detection Method: | |
| Confidence: | 166.0 |
| Match: | 1iwoA |
| Description: | Calcium ATPase, transduction domain A; Calcium ATPase, catalytic domain P; Calcium ATPase; Calcium ATPase, transmembrane domain M |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [1103-1211] |
1 11 21 31 41 51
| | | | | |
1 LNTAIYLLQL IQQVSTFAIN YQGRPFREAL SENKGMYYGL LGIAFVAIAG VTEFSPELNA 60
61 KLQLVKMAYN FQIQLLATMV VDYAACWIIE ELMKKYFRDN KPKEIVLRN
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
| MCM Score |
GO Score |
GO Term |
SCOP Match |
SCOP Description | ||
| View | Download | 0.889 | 0.000 | transport | a.7.1 | Spectrin repeat |