| Protein: | CG32669-PA |
| Organism: | Drosophila melanogaster |
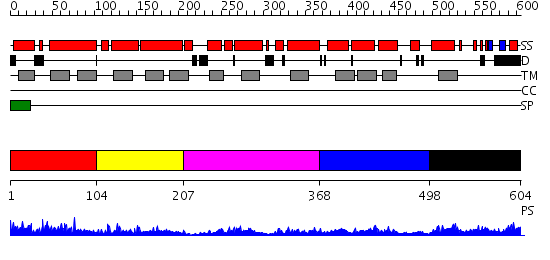
| Length: | 604 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for CG32669-PA.
| Description | E-value | Query Range |
Subject Range |
|
|
365.0 | [0..2] | [573..7] |
|
Region A: Residues: [1-103] |
1 11 21 31 41 51
| | | | | |
1 MATLGAWDYT ILAVVLIISV AIGIYYRFVG GKQSTTTEYL LADRSMNVAP VAFSLMASFM 60
61 SAVTILGVSM ENYQYGTMFV VINLSYVLST PVAAYLIIPV FYR
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [104-206] |
1 11 21 31 41 51
| | | | | |
1 LKTASVYEYL ELRFGYATRL AASLSFSLQM VLYMGIVVYA PALALEAVTG LSQVFSIVIV 60
61 GVVCTFYATL GGMKAVLITD IYQSLLMFAA VFSVIICAWV KAG
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [207-367] |
1 11 21 31 41 51
| | | | | |
1 SLEVIWRVAQ ENGRINLTNF SVDPTERHTW FTQILGGCAT YLAIYGVNQT QVQRLMAVKS 60
61 LSAARAALWW CLPILCLLSL STCFSGLCIY WYYRDCDPLL EGRVNSRDQV MPLFVVDTMG 120
121 EYTGLAGLFV SGIFCASLST ISSIISSLAA VTLEDYLKPL V
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [368-497] |
1 11 21 31 41 51
| | | | | |
1 SCCAKRTLTD RQTLWYSKLL SLFFGALCIG MAFMAGSIGG LLQAALSIFG IIGGPLLGLF 60
61 TLGMYVTKAN EKGAIGGLLI SLAFCFWIGF GQPKPPLVSL DMSTAGCPVD RSFARDIFLK 120
121 TVIAVAEEEH
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [498-604] |
1 11 21 31 41 51
| | | | | |
1 YFYLYRISYM WYAALGFLIT FFGGWLLSWL FALLKWDNNR RIYQDADCTL IKHDLFVPPI 60
61 AKRLQRRQMP LLVVTGTSSE IGGITTESAT APPRLDEIEW EKHKAVA
|
| Detection Method: | |
| Confidence: | 1.23 |
| Match: | 4hb1A |
| Description: | A DESIGNED FOUR HELIX BUNDLE PROTEIN. |
Matching Structure (courtesy of the PDB): |
|