| Protein: | gi|24646188, gi|... |
| Organism: | Drosophila melanogaster |
| Length: | 1718 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for gi|24646188, gi|....
| Description | E-value | Query Range |
Subject Range |
|
|
1072.0 | [0..3] | [1718..1] |
|
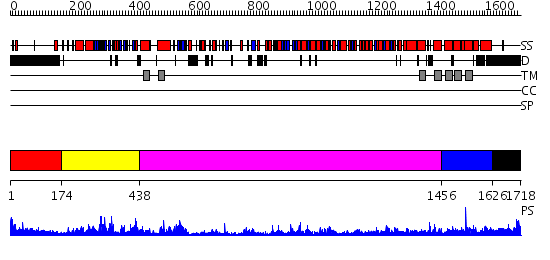
Region A: Residues: [1-173] |
1 11 21 31 41 51
| | | | | |
1 MIGGRRCSEA ARRLRRRPDT LELSVLGGQP IETELQEDNE RTPTKGAGNV DEPGIEAPEA 60
61 SCSAPSKRTR WFQFARSSKN RRKRLHCNED EDVKQPERRN SDLEGSPRAC LAQPEGMNIG 120
121 SSTQTIATPL MIGDSFYSLA PGGGNPPPNK SDDQELYKQR DPSVTSESKS VPS
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [174-437] |
1 11 21 31 41 51
| | | | | |
1 LRRLSQIRRR RSSYYFSENE RRIRANDKEF NAQFKYHNNY IKTSKYSLFT FLPFNLLEQF 60
61 QRLANFYFLC LLVLQLIPAI SSLTPVTTAI PLIGVLTLTA VKDAYDDIVN NRKSKTLRNG 120
121 KLVEAKWSEV QVGDVIRLDN NQFVAADTLL LSTSEPNGLC FIETAELDGE TNLKAKQCLT 180
181 ETIELGDRHD SLWNFNGEII CERPNNLLNK FDGTLIWRGQ RFALDNEKIL LRGCVLRNTQ 240
241 WCYGVVVFAG VDTKLMQNSG KTQF
|
| Detection Method: | |
| Confidence: | 39.221849 |
| Match: | 1mhsA |
| Description: | Proton ATPase |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [438-1455] |
1 11 21 31 41 51
| | | | | |
1 KSTGVDRLLN FIIIGIVLFL VSICALFAIG CAIWEGLIGQ HFQLYLPWEH IIPKDYIPTG 60
61 ATVIGLLVFF SYAIVLNTVV PISLYVSVEV IRFVQSFLIN WDEEMYYPTT NTYAKARTTT 120
121 LNEELGQIQY IFSDKTGTLT QNIMTFNKCS INGRSYGDVI DLRTGELVEI TEQQTIFQNS 180
181 NTNNRPSPLS GAIVAPPAAP PPIILVHKAE VHAKKTSMVV TSSGEAQVLP DRPRSDIERS 240
241 APPMDASEKR PGLKHVRYSA PSRSQDADAG RLSPRLDGSG GLSPPVGNEE RRISGGFKRS 300
301 GAGCMQRQLS RTSSCDKVKI LHDSQQTETH NMHHSSHNRK RLVKIRFKKA PSTATLGVVI 360
361 SQLEHHLDEE PQSTSTLSTT KHHHHRSKAF ATNWSSSPHR VHALQSVDFS ANPHHESDFR 420
421 WYDRTLLDAV RSDEEHSHVF FRLLALCHTV MAETVDGKLE YQAQSPDEAA LVSAARNFGF 480
481 VFRTRTPNSI TIEVMGQTEE YELLNILDFN NVRKRMSVIL RRGDSMVLYC KGADNVIYDR 540
541 LHGGQEDLKA RTQDHLNKFA GEGLRTLALA ERRLTEQYYN DWRSRQQEAA LSMDSREQKL 600
601 NAIYEEIESE MQLVGVTAIE DKLQDGVPKS IANLQNAGIK IWVLTGDKQE TAINIGYSCQ 660
661 LLTDELADVF IVDGNSVEEV EKQLRQFKES IKIYNRFRPG GFDPFDRLNS DSNMDPLSVT 720
721 MTQTSAFMQE TNLPPTPPPP PAISVVTFRW DEKNKDNKGG PDSAECNNLF GDEKGSEDGG 780
781 TASIVVDENT GFALVVNGHS LVHCLSPELE NKFLDIASQC KAVICCRVTP LQKALVVELI 840
841 KRAKNAVTLA IGDGANDVSM IKAAHIGVGI SGQEGLQAVL SSDYSIAQFR YLERLLLVHG 900
901 RWSYYRMCKF LRYFFYKNFA FTLCHCWYSL FCGFSAQTVF DPMFISVYNL FYTSLPVLAL 960
961 GVFEQDVSDK NSLEFPRLYT PGLKSELFNI REFIYSVLHG AFTSLVLFLI PYGVYKDG
|
| Detection Method: | |
| Confidence: | 88.39794 |
| Match: | 1iwoA |
| Description: | Calcium ATPase, transduction domain A; Calcium ATPase, catalytic domain P; Calcium ATPase; Calcium ATPase, transmembrane domain M |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [1456-1625] |
1 11 21 31 41 51
| | | | | |
1 VSANGFVVSD HMTLGAVVAT ILIVDNTAQI SLYTSYWTVV NHVTIWGSLV WYFVLDYFYN 60
61 YVIGGPYVGS LTQAMKDLTF WVTMLITVMA LVAPVLAYKF YLLDVHPSLS DKIRQKSLKK 120
121 IHSRASSDVR RTASSRRGRR SVRSGYAFAH QEGFGRLITS GKIMHKLPQD
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [1626-1718] |
1 11 21 31 41 51
| | | | | |
1 FAFPLGLGTK KTQVLHNNLN SADGPNSKGN NVTGHHMVNN NTNMRHNQNQ NHSSMADITA 60
61 DGRGNGGDDG RGSGGSDDMS PRAPCQDLDT INL
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.