| Protein: | disp-PA |
| Organism: | Drosophila melanogaster |
| Length: | 1218 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for disp-PA.
| Description | E-value | Query Range |
Subject Range |
|
|
802.0 | [0..1] | [1218..1] |
|
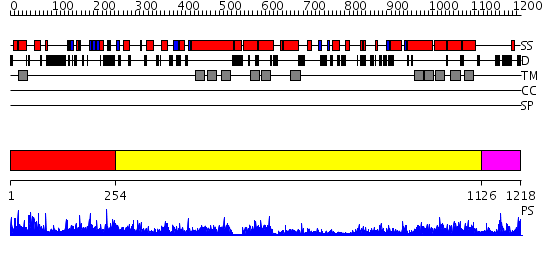
Region A: Residues: [1-253] |
1 11 21 31 41 51
| | | | | |
1 MLCFDSERMN WYYHVLARRP YLVVVSIAVY CVACIIVALV LNKLPDFSDP TLGFETRGTK 60
61 IGERLTAWYN LLQETDHHGA LFSNPSDLWE RRRVEQGYVE TKLHPNHRRR KNKHKNRNKN 120
121 KRRKEQNQSS HEHHDVAQKM MQFKKRLKAT SSPSPNLGFD TWIGDSGVFR DYEITNDSAS 180
181 SSLEPTRRTE QIEYGHNTTS VDEEEHQQRV QTKKSTWRLL KQAATLPTDG WADMHRRQPI 240
241 EGFFCDSSPR KEY
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [254-1125] |
1 11 21 31 41 51
| | | | | |
1 SHFVVQRIGP NATDSLFDLN GLLAMCQLQD QITEVPSYRA FCEPEMLTTE CCRPWSLPNY 60
61 AAMLANKSSC FDLTTEDVTS LHTLLLGCYE YFHDLKMDNH CNEIPHCRAP EECKRLNIVF 120
121 NVLNFLTDFS FIKSNDSNVY LKYAMIFIPV AQSNRLLPLF HEWEDVELIN ELVEVVAMDL 180
181 GLENELFNEL LLTDVWLVSL GGTFVMASVW LYTGSAFITL MSCVAICFSL GLAYFFYAIV 240
241 LEFEFFPYMN LLAVVVIIGI GADDVFLFLK IWHCVLTERF SNRCTLTTQS QSALPTLENS 300
301 DHTESLENIM ALTMRHAAAS MFVTSLTTAG AFYASYSSSI TAIKCFGIFA GTVVVTNYLL 360
361 MITWLPASVS IMERLFATRM SCHHPMSIKL IHACKKSINR FCQMFEECIT KSIMNYAYLW 420
421 LLIFGALGAS SAVIVFWYPG LQLPEKSHFQ LFVSKHPFEV YSSLKQQFWF EKPLQAYENF 480
481 KMHMHFVWGV QAVDDGDYTN PNSYGHLHYD NNFNVSSRPA QLWILDFCQS VRQQPFYKET 540
541 LGMLLPNCFI ENLIDYMKRR CIDDMDSTRK DRSPCCDAQF PFEPHIFEYC LPQSISNMYD 600
601 TTFFRPGVAG PKFAEAPRLE TEDYLGMSGN ESAEYSTNGS FTPLLVKALV IEFESNVAYS 660
661 TIYANIRQFY ESVEHWFQMQ LKTAPPELQG GWFTSDLKFY NVQDTLSHDT FVAICLAMAA 720
721 SLAVLLCFTV NILISIYAVL TVSLSIFNTV AVLILLGWQL NILESIAVST AIGLAVDFSL 780
781 HYGIHYRMSP VKERLAATQF VLSRIIGPTV MAATTTGLAG GIMMASNILP YIQIGVFLVV 840
841 VMIVSWFYAT FFLMSLLRVA GPQHGFLELK WP
|
| Detection Method: | |
| Confidence: | 177.0 |
| Match: | 1oy6A |
| Description: | Multidrug efflux transporter AcrB pore domain; PN1, PN2, PC1 and PC2 subdomains; Multidrug efflux transporter AcrB TolC docking domain; DN and DC subdomains; Multidrug efflux transporter AcrB transmembrane domain |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [1126-1218] |
1 11 21 31 41 51
| | | | | |
1 LWSKRSSGSS KFYERKPSQV IASEQLLTPT SSAIVELANS ETHELESLNS NSLIKTISGI 60
61 ESAHALSSLP RDFEHSFQTM HECKYQTYPS TSN
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.