| Protein: | CG30116-PC, CG30... |
| Organism: | Drosophila melanogaster |
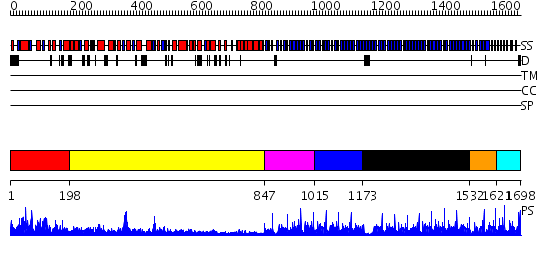
| Length: | 1698 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for CG30116-PC, CG30....
| Description | E-value | Query Range |
Subject Range |
|
|
1252.0 | [0..1] | [1694..1] |
|
Region A: Residues: [1-197] |
1 11 21 31 41 51
| | | | | |
1 MAAPIDESVL NALRGVTSAH TRLPKPLLIK IYVASLKQEF NQERRMLLEL VGPELQSLYD 60
61 DRQIELEFVD MHFGTGDLEV HQLERDPYLI HDYLHEIDTC HAHTKSVFFM VLVGDGIGRQ 120
121 LLPTKIDEDI FSAVLADQQT SADHEAMLVK WYERDASQTQ RQLKQDYRLM NVDAWLAESQ 180
181 RMQSFLEQAF QSLLQGS
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [198-846] |
1 11 21 31 41 51
| | | | | |
1 GASGRSPDFM ERVQLLRRTQ IEREVSQAMA LTSEKILAVF RERAAQCSKG DVEAAERLRK 60
61 IKDELTMNLS TDNHTTLVVP GSAASEAIDP DNEDHESYLS KFKNKVTDKL RLLIEAHITN 120
121 DPDVIKGRKK TVQEIFHEHA THLRILREHT DSDALVESRV PQQLRQNLMA NFRNGSRHAP 180
181 YFLCGADGSG KSAILCHLYG QVGSWFGSTR VHRVIRFAKA TPRSAYNLEL LRVICQQISI 240
241 IFNIPEGYLP KDASFDPLYI NTWFQNLLRR VEDMGNDVLF LFIDDLHLLN PLDCDIVTAL 300
301 SWLPTSLPWN VQIICSSTTP VEQLKFTPMQ RDRFKSIEYQ FDLNMGDYAT KLKIPPQCIP 360
361 GDVSFALYVE QQFDQLERHY GRQAVGDLAS YITCSEYGLS ETELLELLMP TDDPESLIET 420
421 KNGHFSFATF KKIHREMDLL LLLHDKIMSG KVLIQWRHNY CASVAKRRYM DVQRTRSLHC 480
481 ELANLFFPQD EDESTLENES NRSESKSVIS LKDRDREKDS LSAVSAVSAG RKSSSTHHND 540
541 DTSTFYNPIA ADVSYSMRHV EESWHHLMRS DDTTRFKQIA VCNFDFLLAA VQTVSISYLR 600
601 CLIEHVRCYI LDRDIELIYY TIRKSSDVLT RDPMQLGSQL ISWLRPISE
|
| Detection Method: | |
| Confidence: | 81.0 |
| Match: | 1z6tA |
| Description: | Structure of the apoptotic protease-activating factor 1 bound to ADP |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [847-1014] |
1 11 21 31 41 51
| | | | | |
1 HDDDDNSLLS MTVRSATAWC DGYAVPLLVP LTGWLPAPLP SQIRTMTVSG TGCIRAVWLA 60
61 PSKQHLILAT SSGDVQQWHI MSNSLDHIFK GHTAAVTCLL VAPQSDSELL LTGSEDATVL 120
121 VWHVGLRERR AHIKNAHTAP ITGVAAGANN TLIISSSEDA SIAITDLA
|
| Detection Method: | |
| Confidence: | 70.045757 |
| Match: | 2ovpB |
| Description: | No description for 2ovpB was found. |
|
Region A: Residues: [1015-1172] |
1 11 21 31 41 51
| | | | | |
1 SGKLRHRITH HRGPVSGIVV AGACDVLISG GLDRTICVWD LDNFTLLNTM QMTSAVLRID 60
61 ISWNSVFLLA LCEDNALYVR TLATGKELHT LKGHKSKIRS ISIGKDSQRC VVGCDDTRAL 120
121 IYDMHAGKLV RSLPPNPGPV TAVHAMDNDD FLVTVGGN
|
| Detection Method: | |
| Confidence: | 4.30103 |
| Match: | 1r9nA |
| Description: | Crystal Structure of human dipeptidyl peptidase IV in complex with a decapeptide (tNPY) at 2.3 Ang. Resolution |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [1173-1531] |
1 11 21 31 41 51
| | | | | |
1 KITFYSFRNE ELYVNPYSRH PRRKRSLKRH AQAQRSPSTT LPPITCFDLS RDSQQMAIAS 60
61 GRHVHLMRIN TPEYQCTLEG HTAGVSCLKF APNGEFLATG SEDRLVHIWN LALGEICNSF 120
121 KGHTAPVVKV VVLMDSLRVI STDRDSMLLV WMAHSGNLLQ TIQGPYKSLS VTNNMRFAVS 180
181 TNGDNTLKIW SLTQEDEKYS VSHSDEITCF EISADSVHII SGSRDMSLKV WQATGGKLSQ 240
241 VLVGHSDAVT CVAVSVTNKT QVLSGSKDMN LILWDLLTGE EVHTLAGHLG PVIGVKVSAD 300
301 GSTAVSGSDD KTLIVWETKR GLALTSLQMH VPFTHFDISL EVSRVLVQLV DSYNLPVIC
|
| Detection Method: | |
| Confidence: | 49.0 |
| Match: | 1nexB |
| Description: | Cdc4 F-box and linker domains; Cdc4 propeller domain |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [1532-1620] |
1 11 21 31 41 51
| | | | | |
1 LHNTPAQYVK LPTYSGPSKD VEDLRPQGPK RQMKRLLKKE VSLDTYTWQK KYGHLTSSVM 60
61 MAQVDERLKR RFSVSASMEE ISKIAETKT
|
| Detection Method: | |
| Confidence: | 54.69897 |
| Match: | 1trjA |
| Description: | Homology Model of Yeast RACK1 Protein fitted into 11.7A cryo-EM map of Yeast 80S Ribosome |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [1621-1698] |
1 11 21 31 41 51
| | | | | |
1 GASQANLGPE QAALAQSQHF DQLEALWNKR SPPRRRHNAG LSRQTSLVED RLESSDDDEY 60
61 QDERADHFAS GGGGYHKG
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.