| Protein: | SULH2_SCHPO |
| Organism: | Schizosaccharomyces pombe 972h- |
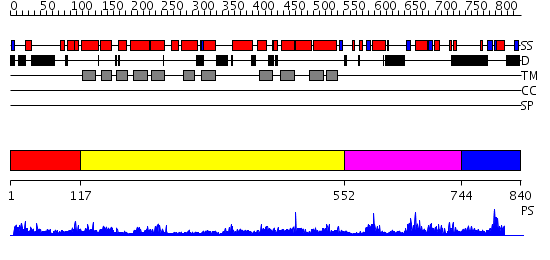
| Length: | 840 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for SULH2_SCHPO.
| Description | E-value | Query Range |
Subject Range |
|
|
533.0 | [0..70] | [814..83] |
|
|
514.0 | [0..56] | [808..2] |
|
|
494.0 | [0..57] | [808..31] |
|
|
493.0 | [0..67] | [808..45] |
|
|
479.0 | [0..71] | [808..46] |
|
|
476.0 | [0..77] | [808..129] |
|
|
474.0 | [0..71] | [808..46] |
|
|
470.0 | [0..47] | [808..59] |
|
|
464.0 | [0..69] | [808..37] |
|
Region A: Residues: [1-116] |
1 11 21 31 41 51
| | | | | |
1 MRAWGWVRNK FSSEDDYNDG ASNKDYPDRF NEFDNSQNDH NDYTQNNAQF QNAQTTTFGR 60
61 TISRVKAYYE IPEDDELDEL ASIPQWFKKN VTSNIFKNFL HYLKSLFPII EWLPNY
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [117-551] |
1 11 21 31 41 51
| | | | | |
1 NPYWLINDLI AGITVGCVVV PQGMSYAKVA TLPSEYGLYS SFVGVAIYCF FATSKDVSIG 60
61 PVAVMSLITA KVIANVMAKD ETYTAPQIAT CLALLAGAIT CGIGLLRLGF IIEFIPVPAV 120
121 AGFTTGSALN ILSGQVPALM GYKNKVTAKA TYMVIIQSLK HLPDTTVDAA FGLVSLFILF 180
181 FTKYMCQYLG KRYPRWQQAF FLTNTLRSAV VVIVGTAISY AICKHHRSDP PISIIKTVPR 240
241 GFQHVGVPLI TKKLCRDLAS ELPVSVIVLL LEHISIAKSF GRVNDYRIVP DQELIAMGVT 300
301 NLIGIFFNAY PATGSFSRSA IKAKAGVKTP IAGIFTAAVV ILSLYCLTDA FYYIPNAILS 360
361 AVIIHAVTDL ILPMKQTILF WRLQPLEACI FFISVIVSVF SSIENGIYVS VCLAAALLLL 420
421 RIAKPHGSFL GKIQA
|
| Detection Method: | |
| Confidence: | 0.988 |
| Match: | 2a65A |
| Description: | Crystal structure of LEUTAA, a bacterial homolog of Na+/Cl--dependent neurotransmitter transporters |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [552-743] |
1 11 21 31 41 51
| | | | | |
1 ANKYGSDNIA NVRDIYVPLE MKEENPNLEI QSPPPGVFIF RLQESFTYPN ASRVSTMISR 60
61 RIKDLTRRGI DNIYVKDIDR PWNVPRQRKK KENSEIEDLR PLLQAIIFDF SAVNNLDTTA 120
121 VQSLIDIRKE LEIYANETVE FHFTNIRSGW IKRTLVAAGF GKPKGHAVDA SVCVEVAAPL 180
181 RDANLSAESS RN
|
| Detection Method: | |
| Confidence: | 1.51 |
| Match: | 1auzA |
| Description: | SOLUTION STRUCTURE OF SPOIIAA, A PHOSPHORYLATABLE COMPONENT OF THE SYSTEM THAT REGULATES TRANSCRIPTION FACTOR SIGMA-F OF BACILLUS SUBTILIS, NMR, 24 STRUCTURES |
Matching Structure (courtesy of the PDB): |
|
| Term | Confidence | Notes |
| binding | 2.14643407404573 | bayes_pls_golite062009 |
| protein binding | 1.48694301635456 | bayes_pls_golite062009 |
|
Region A: Residues: [744-840] |
1 11 21 31 41 51
| | | | | |
1 LSRIITPIYD DEEGNVSGHI YELDGKNNSD LSMHCQKGSN QVEIEFVEFN SRKYPFFHVD 60
61 VASAVVDLQH RLLSPQKSDS FGSLKEGGTT TIKKIEN
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
| MCM Score |
GO Score |
GO Term |
SCOP Match |
SCOP Description | ||
| View | Download | 0.607 | 0.823 | secondary active sulfate transmembrane transporter activity | b.1.1 | Immunoglobulin |