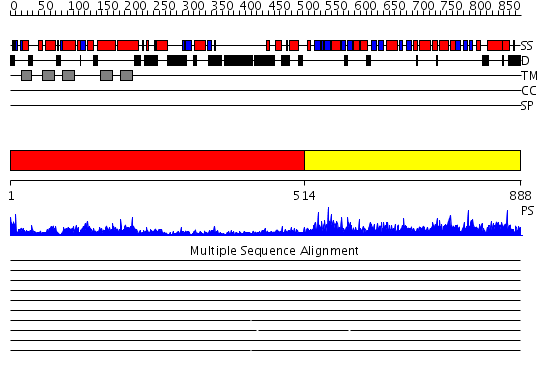
| Protein: | HMGCR |
| Organism: | Homo sapiens |
| Length: | 888 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for HMGCR.
| Description | E-value | Query Range |
Subject Range |
|
|
0.0 | [1..888] | [1..888] |
|
|
0.0 | [1..888] | [1..888] |
|
|
0.0 | [1..888] | [1..888] |
|
|
0.0 | [1..888] | [91..978] |
|
|
0.0 | [1..888] | [1..888] |
|
|
0.0 | [1..888] | [1..888] |
|
|
0.0 | [1..888] | [1..887] |
|
|
0.0 | [1..888] | [1..885] |
|
|
0.0 | [1..888] | [1..887] |
|
Region A: Residues: [1-513] |
1 11 21 31 41 51
| | | | | |
1 MLSRLFRMHG LFVASHPWEV IVGTVTLTIC MMSMNMFTGN NKICGWNYEC PKFEEDVLSS 60
61 DIIILTITRC IAILYIYFQF QNLRQLGSKY ILGIAGLFTI FSSFVFSTVV IHFLDKELTG 120
121 LNEALPFFLL LIDLSRASTL AKFALSSNSQ DEVRENIARG MAILGPTFTL DALVECLVIG 180
181 VGTMSGVRQL EIMCCFGCMS VLANYFVFMT FFPACVSLVL ELSRESREGR PIWQLSHFAR 240
241 VLEEEENKPN PVTQRVKMIM SLGLVLVHAH SRWIADPSPQ NSTADTSKVS LGLDENVSKR 300
301 IEPSVSLWQF YLSKMISMDI EQVITLSLAL LLAVKYIFFE QTETESTLSL KNPITSPVVT 360
361 QKKVPDNCCR REPMLVRNNQ KCDSVEEETG INRERKVEVI KPLVAETDTP NRATFVVGNS 420
421 SLLDTSSVLV TQEPEIELPR EPRPNEECLQ ILGNAEKGAK FLSDAEIIQL VNAKHIPAYK 480
481 LETLMETHER GVSIRRQLLS KKLSEPSSLQ YLP
|
| Detection Method: | |
| Confidence: | 71.522879 |
| Match: | 1oy6A |
| Description: | Multidrug efflux transporter AcrB pore domain; PN1, PN2, PC1 and PC2 subdomains; Multidrug efflux transporter AcrB TolC docking domain; DN and DC subdomains; Multidrug efflux transporter AcrB transmembrane domain |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [514-888] |
1 11 21 31 41 51
| | | | | |
1 YRDYNYSLVM GACCENVIGY MPIPVGVAGP LCLDEKEFQV PMATTEGCLV ASTNRGCRAI 60
61 GLGGGASSRV LADGMTRGPV VRLPRACDSA EVKAWLETSE GFAVIKEAFD STSRFARLQK 120
121 LHTSIAGRNL YIRFQSRSGD AMGMNMISKG TEKALSKLHE YFPEMQILAV SGNYCTDKKP 180
181 AAINWIEGRG KSVVCEAVIP AKVVREVLKT TTEAMIEVNI NKNLVGSAMA GSIGGYNAHA 240
241 ANIVTAIYIA CGQDAAQNVG SSNCITLMEA SGPTNEDLYI SCTMPSIEIG TVGGGTNLLP 300
301 QQACLQMLGV QGACKDNPGE NARQLARIVC GTVMAGELSL MAALAAGHLV KSHMIHNRSK 360
361 INLQDLQGAC TKKTA
|
| Detection Method: | |
| Confidence: | 1000.0 |
| Match: | 1dq8A |
| Description: | NAD-binding domain of HMG-CoA reductase; Substrate-binding domain of HMG-CoA reductase |
Matching Structure (courtesy of the PDB): |
|