| Protein: | KDR |
| Organism: | Homo sapiens |
| Length: | 1356 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for KDR.
| Description | E-value | Query Range |
Subject Range |
|
|
0.0 | [1..1356] | [1..1356] |
|
|
0.0 | [1..1356] | [111..1466] |
|
|
0.0 | [1..1356] | [111..1466] |
|
|
0.0 | [1..1356] | [1..1355] |
|
|
0.0 | [1..1356] | [1..1345] |
|
|
0.0 | [1..1356] | [1..1343] |
|
|
0.0 | [1..1356] | [1..1345] |
|
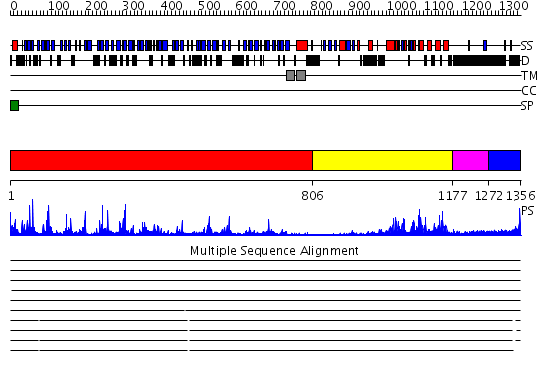
Region A: Residues: [1-805] |
1 11 21 31 41 51
| | | | | |
1 MQSKVLLAVA LWLCVETRAA SVGLPSVSLD LPRLSIQKDI LTIKANTTLQ ITCRGQRDLD 60
61 WLWPNNQSGS EQRVEVTECS DGLFCKTLTI PKVIGNDTGA YKCFYRETDL ASVIYVYVQD 120
121 YRSPFIASVS DQHGVVYITE NKNKTVVIPC LGSISNLNVS LCARYPEKRF VPDGNRISWD 180
181 SKKGFTIPSY MISYAGMVFC EAKINDESYQ SIMYIVVVVG YRIYDVVLSP SHGIELSVGE 240
241 KLVLNCTART ELNVGIDFNW EYPSSKHQHK KLVNRDLKTQ SGSEMKKFLS TLTIDGVTRS 300
301 DQGLYTCAAS SGLMTKKNST FVRVHEKPFV AFGSGMESLV EATVGERVRI PAKYLGYPPP 360
361 EIKWYKNGIP LESNHTIKAG HVLTIMEVSE RDTGNYTVIL TNPISKEKQS HVVSLVVYVP 420
421 PQIGEKSLIS PVDSYQYGTT QTLTCTVYAI PPPHHIHWYW QLEEECANEP SQAVSVTNPY 480
481 PCEEWRSVED FQGGNKIEVN KNQFALIEGK NKTVSTLVIQ AANVSALYKC EAVNKVGRGE 540
541 RVISFHVTRG PEITLQPDMQ PTEQESVSLW CTADRSTFEN LTWYKLGPQP LPIHVGELPT 600
601 PVCKNLDTLW KLNATMFSNS TNDILIMELK NASLQDQGDY VCLAQDRKTK KRHCVVRQLT 660
661 VLERVAPTIT GNLENQTTSI GESIEVSCTA SGNPPPQIMW FKDNETLVED SGIVLKDGNR 720
721 NLTIRRVRKE DEGLYTCQAC SVLGCAKVEA FFIIEGAQEK TNLEIIILVG TAVIAMFFWL 780
781 LLVIILRTVK RANGGELKTG YLSIV
|
| Detection Method: | |
| Confidence: | 36.09691 |
| Match: | 1e07A |
| Description: | Model of human carcinoembryonic antigen by homology modelling and curve-fitting to experimental solution scattering data |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [806-1176] |
1 11 21 31 41 51
| | | | | |
1 MDPDELPLDE HCERLPYDAS KWEFPRDRLK LGKPLGRGAF GQVIEADAFG IDKTATCRTV 60
61 AVKMLKEGAT HSEHRALMSE LKILIHIGHH LNVVNLLGAC TKPGGPLMVI VEFCKFGNLS 120
121 TYLRSKRNEF VPYKTKGARF RQGKDYVGAI PVDLKRRLDS ITSSQSSASS GFVEEKSLSD 180
181 VEEEEAPEDL YKDFLTLEHL ICYSFQVAKG MEFLASRKCI HRDLAARNIL LSEKNVVKIC 240
241 DFGLARDIYK DPDYVRKGDA RLPLKWMAPE TIFDRVYTIQ SDVWSFGVLL WEIFSLGASP 300
301 YPGVKIDEEF CRRLKEGTRM RAPDYTTPEM YQTMLDCWHG EPSQRPTFSE LVEHLGNLLQ 360
361 ANAQQDGKDY I
|
| Detection Method: | |
| Confidence: | 61.0 |
| Match: | 1y6aA |
| Description: | Crystal structure of VEGFR2 in complex with a 2-anilino-5-aryl-oxazole inhibitor |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [1177-1271] |
1 11 21 31 41 51
| | | | | |
1 VLPISETLSM EEDSGLSLPT SPVSCMEEEE VCDPKFHYDN TAGISQYLQN SKRKSRPVSV 60
61 KTFEDIPLEE PEVKVIPDDN QTDSGMVLAS EELKT
|
| Detection Method: | |
| Confidence: | 60.30103 |
| Match: | 1mrvA |
| Description: | crystal structure of an inactive Akt2 kinase domain |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [1272-1356] |
1 11 21 31 41 51
| | | | | |
1 LEDRTKLSPS FGGMVPSKSR ESVASEGSNQ TSGYQSGYHS DDTDTTVYSS EEAELLKLIE 60
61 IGVQTGSTAQ ILQPDSGTTL SSPPV
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.