| Protein: | APE2 |
| Organism: | Saccharomyces cerevisiae |
| Length: | 935 amino acids |
| Reference: | Malmström L, et al. (2007) Superfamily assignments for the yeast proteome through integration of structure prediction with the gene ontology. PLoS Biol 5(4): e76. doi:10.1371/journal.pbio.0050076 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for APE2.
| Description | E-value | Query Range |
Subject Range |
|
|
0.0 | [1..935] | [1..935] |
|
|
0.0 | [16..934] | [1..929] |
|
|
0.0 | [102..934] | [17..847] |
|
|
0.0 | [16..934] | [1..929] |
|
|
0.0 | [102..934] | [22..859] |
|
|
0.0 | [83..934] | [2..859] |
|
|
0.0 | [31..934] | [8..950] |
|
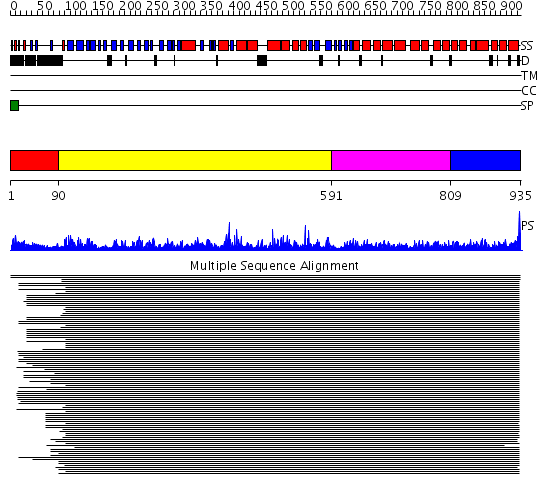
Region A: Residues: [1-89] |
1 11 21 31 41 51
| | | | | |
1 MPIVRWLLLK SAVRGSSLIG KAHPCLRSIA AHPRYLSNVY SPPAGVSRSL RINVMWKQSK 60
61 LTPPRFVKIM NRRPLFTETS HACAKCQKT
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [90-590] |
1 11 21 31 41 51
| | | | | |
1 SQLLNKTPNR EILPDNVVPL HYDLTVEPDF KTFKFEGSVK IELKINNPAI DTVTLNTVDT 60
61 DIHSAKIGDV TSSEIISEEE QQVTTFAFPK GTMSSFKGNA FLDIKFTGIL NDNMAGFYRA 120
121 KYEDKLTGET KYMATTQMEP TDARRAFPCF DEPNLKASFA ITLVSDPSLT HLSNMDVKNE 180
181 YVKDGKKVTL FNTTPKMSTY LVAFIVAELK YVESKNFRIP VRVYATPGNE KHGQFAADLT 240
241 AKTLAFFEKT FGIQYPLPKM DNVAVHEFSA GAMENWGLVT YRVVDLLLDK DNSTLDRIQR 300
301 VAEVVQHELA HQWFGNLVTM DWWEGLWLNE GFATWMSWYS CNEFQPEWKV WEQYVTDTLQ 360
361 HALSLDSLRS SHPIEVPVKK ADEINQIFDA ISYSKGASLL RMISKWLGEE TFIKGVSQYL 420
421 NKFKYGNAKT EDLWDALADA SGKDVRSVMN IWTKKVGFPV ISVSEDGNGK ITFRQNRYLS 480
481 TADVKPDEDK TIYPVFLALK T
|
| Detection Method: | |
| Confidence: | 84.69897 |
| Match: | 1gw6A_ |
| Description: | Leukotriene A4 hydrolase C-terminal domain; Leukotriene A4 hydrolase N-terminal domain; Leukotriene A4 hydrolase catalytic domain |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [591-808] |
1 11 21 31 41 51
| | | | | |
1 KNGVDSSVVL SERSKTIELE DPTFFKVNSE QSGIYITSYT DERWAKLGQQ ADLLSVEDRV 60
61 GLVADVKTLS ASGYTSTTNF LNLVSKWNNE KSFVVWDQII NSISSMKSTW LFEPKETQDA 120
121 LDNFTKQLIS GMTHHLGWEF KSSDSFSTQR LKVTMFGAAC AARDADVEKA ALKMFTDYCS 180
181 GNKEAIPALI KPIVFNTVAR VGGAENYEKV YKIYLDPI
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [809-935] |
1 11 21 31 41 51
| | | | | |
1 SNDEKLAALR SLGRFKEPKL LERTLGYLFD GTVLNQDIYI PMQGMRAHQE GVEALWNWVK 60
61 KNWDELVKRL PPGLSMLGSV VTLGTSGFTS MQKIDEIKKF FATKSTKGFD QSLAQSLDTI 120
121 TSKAQWG
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.