| Protein: | STV1_YEAST |
| Organism: | Saccharomyces cerevisiae S288c |
| Length: | 890 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for STV1_YEAST.
| Description | E-value | Query Range |
Subject Range |
|
|
0.0 | [7..890] | [4..843] |
|
|
0.0 | [1..890] | [1..890] |
|
|
0.0 | [6..882] | [3..839] |
|
|
0.0 | [4..889] | [13..889] |
|
|
0.0 | [6..887] | [3..844] |
|
|
0.0 | [6..890] | [3..835] |
|
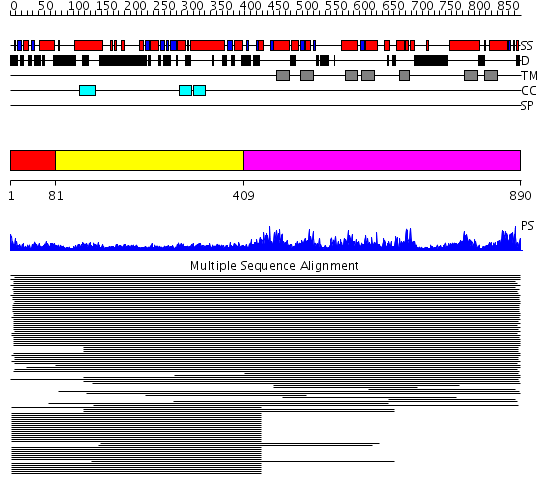
Region A: Residues: [1-80] |
1 11 21 31 41 51
| | | | | |
1 MNQEEAIFRS ADMTYVQLYI PLEVIREVTF LLGKMSVFMV MDLNKDLTAF QRGYVNQLRR 60
61 FDEVERMVGF LNEVVEKHAA
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
| MCM Score |
GO Score |
GO Term |
SCOP Match |
SCOP Description | ||
| View | Download | 0.814 | 0.459 | vacuolar acidification | a.39.1 | EF-hand |
|
Region A: Residues: [81-408] |
1 11 21 31 41 51
| | | | | |
1 ETWKYILHID DEGNDIAQPD MADLINTMEP LSLENVNDMV KEITDCESRA RQLDESLDSL 60
61 RSKLNDLLEQ RQVIFECSKF IEVNPGIAGR ATNPEIEQEE RDVDEFRMTP DDISETLSDA 120
121 FSFDDETPQD RGALGNDLTR NQSVEDLSFL EQGYQHRYMI TGSIRRTKVD ILNRILWRLL 180
181 RGNLIFQNFP IEEPLLEGKE KVEKDCFIIF THGETLLKKV KRVIDSLNGK IVSLNTRSSE 240
241 LVDTLNRQID DLQRILDTTE QTLHTELLVI HDQLPVWSAM TKREKYVYTT LNKFQQESQG 300
301 LIAEGWVPST ELIHLQDSLK DYIETLGS
|
| Detection Method: | |
| Confidence: | 22.221849 |
| Match: | 1i84S_ |
| Description: | Heavy meromyosin subfragment |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [409-890] |
1 11 21 31 41 51
| | | | | |
1 EYSTVFNVIL TNKLPPTYHR TNKFTQAFQS IVDAYGIATY KEINAGLATV VTFPFMFAIM 60
61 FGDMGHGFIL FLMALFLVLN ERKFGAMHRD EIFDMAFTGR YVLLLMGAFS VYTGLLYNDI 120
121 FSKSMTIFKS GWQWPSTFRK GESIEAKKTG VYPFGLDFAW HGTDNGLLFS NSYKMKLSIL 180
181 MGYAHMTYSF MFSYINYRAK NSKVDIIGNF IPGLVFMQSI FGYLSWAIVY KWSKDWIKDD 240
241 KPAPGLLNML INMFLAPGTI DDQLYSGQAK LQVVLLLAAL VCVPWLLLYK PLTLRRLNKN 300
301 GGGGRPHGYQ SVGNIEHEEQ IAQQRHSAEG FQGMIISDVA SVADSINESV GGGEQGPFNF 360
361 GDVMIHQVIH TIEFCLNCIS HTASYLRLWA LSLAHAQLSS VLWDMTISNA FSSKNSGSPL 420
421 AVMKVVFLFA MWFVLTVCIL VFMEGTSAML HALRLHWVEA MSKFFEGEGY AYEPFSFRAI 480
481 IE
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.