| Protein: | GPT1_YEAST |
| Organism: | Saccharomyces cerevisiae S288c |
| Length: | 759 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for GPT1_YEAST.
| Description | E-value | Query Range |
Subject Range |
|
|
0.0 | [1..759] | [1..759] |
|
|
0.0 | [49..749] | [5..672] |
|
|
0.0 | [50..597] | [4..540] |
|
|
7.0E-40 | [119..374] | [4..238] |
|
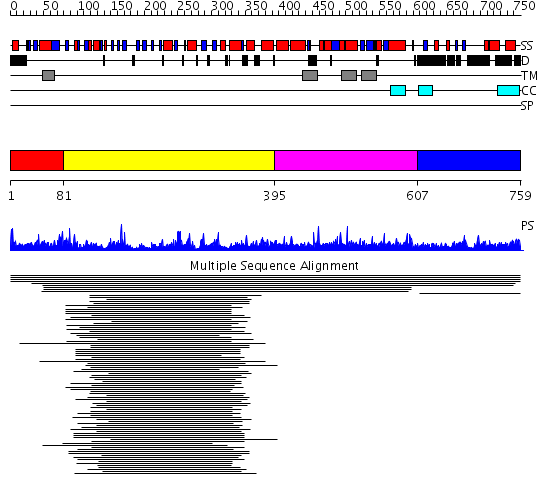
Region A: Residues: [1-80] |
1 11 21 31 41 51
| | | | | |
1 MPAPKLTEKF ASSKSTQKTT NYSSIEAKSV KTSADQAYIY QEPSATKKIL YSIATWLLYN 60
61 IFHCFFREIR GRGSFKVPQQ
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [81-394] |
1 11 21 31 41 51
| | | | | |
1 GPVIFVAAPH ANQFVDPVIL MGEVKKSVNR RVSFLIAESS LKQPPIGFLA SFFMAIGVVR 60
61 PQDNLKPAEG TIRVDPTDYK RVIGHDTHFL TDCMPKGLIG LPKSMGFGEI QSIESDTSLT 120
121 LRKEFKMAKP EIKTALLTGT TYKYAAKVDQ SCVYHRVFEH LAHNNCIGIF PEGGSHDRTN 180
181 LLPLKAGVAI MALGCMDKHP DVNVKIVPCG MNYFHPHKFR SRAVVEFGDP IEIPKELVAK 240
241 YHNPETNRDA VKELLDTISK GLQSVTVTCS DYETLMVVQT IRRLYMTQFS TKLPLPLIVE 300
301 MNRRMVKGYE FYRN
|
| Detection Method: | |
| Confidence: | 25.69897 |
| Match: | 1k30A_ |
| Description: | Glycerol-3-phosphate (1)-acyltransferase |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [395-606] |
1 11 21 31 41 51
| | | | | |
1 DPKIADLTKD IMAYNAALRH YNLPDHLVEE AKVNFAKNLG LVFFRSIGLC ILFSLAMPGI 60
61 IMFSPVFILA KRISQEKART ALSKSTVKIK ANDVIATWKI LIGMGFAPLL YIFWSVLITY 120
121 YLRHKPWNKI YVFSGSYISC VIVTYSALIV GDIGMDGFKS LRPLVLSLTS PKGLQKLQKD 180
181 RRNLAERIIE VVNNFGSELF PDFDSAALRE EF
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [607-759] |
1 11 21 31 41 51
| | | | | |
1 DVIDEEEEDR KTSELNRRKM LRKQKIKRQE KDSSSPIISQ RDNHDAYEHH NQDSDGVSLV 60
61 NSDNSLSNIP LFSSTFHRKS ESSLASTSVA PSSSSEFEVE NEILEEKNGL ASKIAQAVLN 120
121 KRIGENTARE EEEEEEEEEE EEEEEEEGKE GDA
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.