| Protein: | gi|41179262, gi|... |
| Organism: | Lactobacillus prophage Lj965 |
| Length: | 1624 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for gi|41179262, gi|....
| Description | E-value | Query Range |
Subject Range |
|
|
413.0 | [0..306] | [1281..1833] |
|
|
310.0 | [0..315] | [1282..786] |
|
|
294.0 | [0..1224] | [1614..345] |
|
|
259.0 | [0..285] | [1289..959] |
|
|
259.0 | [0..367] | [1280..1121] |
|
|
233.0 | [0..304] | [1281..829] |
|
|
231.0 | [0..302] | [1280..916] |
|
|
230.0 | [0..312] | [1281..846] |
|
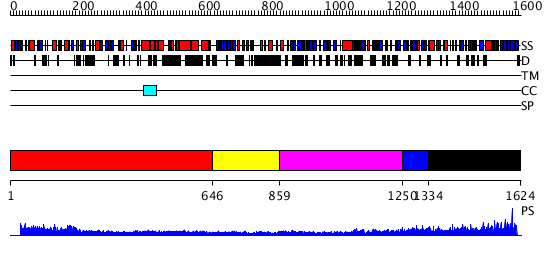
Region A: Residues: [1-645] |
1 11 21 31 41 51
| | | | | |
1 MLTQTKEVRD AWRASQRTLD IKVAVNGKTY TATDINSLKY DSGAYTGDTF AIGSTYSNSV 60
61 QIEFSHLIEN LKLGMEVLPS IGIKTSSGYV YEPLGVFIIS SEIKMDRNNN LTSISASDRF 120
121 CGLEGSYKSK LAYPAKVLDV IAEICEQSGV KANVDDLARL PHQADLPRPI TGQTYRKALG 180
181 WIAQLYAGYA TFDRKGLFTI RTIAEPNYEL DPSQYEQAGL TKNEAPYRIS GIQCQTTITT 240
241 KTRDGEDTDE TKNYQVGDTN GSQIKLENNI MTPDRLTNIW EQIKDVNFYP FSLNWFGNPA 300
301 IEAGDWLKLQ DKQGNKFIVP NNSYTLDFNG GLSATSKADQ TSSTDSVIAW EGTFSQTIRE 360
361 LQGRKAPDGT VIFPPSVTEP PTNAKPNDVW FKQNGNSTEL WVFTEQEDGT RKWIRRDLTP 420
421 DEIKKQVQEA QDGLKDAKKE IADNLAKADK DIAELNESIN NQKGSLDGLS TTVNTVVIPK 480
481 VTDVTNQVSD AIKKVNEQKN IVTGLQNQAI QQGKDISKIT SDVHGVTVDL ANLNGDVNQT 540
541 KATVQGLQST LGNAQGDIAQ IKVDAKKLET SLSGKVDNST YATFVNQTNQ ALNAKLIASD 600
601 LNGYAKTTDL QATANGLQFN INSVTDKLNN LRIGGRNLVG GTDKE
|
| Detection Method: | |
| Confidence: | 1.26 |
| Match: | 2oevA |
| Description: | No description for 2oevA was found. |
|
Region A: Residues: [646-858] |
1 11 21 31 41 51
| | | | | |
1 YVMGFGIPNT VWKDNFAYIS LPLISGDGGE ILPQGAGFWH VLSPGEEYTQ TIWIETDAII 60
61 KSLNGTFLTW LTMDDGHDVQ KAYIQRIGNN SYKIVGSYTW PQNKKGNRVR LFDIFSLPNS 120
121 IDLKSGTYLK FGKLKLEVGN ASTDWTPAVE DTEHDLESLS ARITINSQQF GSYYTKGESD 180
181 NRTNTAKNEV INSIKNDSNW HGLTNILTNS GFI
|
| Detection Method: |
Shown below are all of our de novo (Rosetta) predictions for this domain.
Click here to view only most confident match.
Found no structure predictions for this domain.
|
Region A: Residues: [859-1249] |
1 11 21 31 41 51
| | | | | |
1 QTADGFIQKV QQTMQPIINE NNSGGVNLLT NTYTLRNFNN AGNAGEATGF EIDSGDSHKN 60
61 ELNAGEISTN VFHVYAKDLN HAPVYFGQNF TLPKGTWTLS FLVRHNGSSE QKIRLSFYTD 120
121 DCANRGWIPL GTSDEIDNIW KRHQITFTTT QEIFEQNPRV HMPTDDIVPG GSLYFANFKL 180
181 ESGSIATTWC PAPEDLAKQV AVTELGQTIK GLQSTVSSNY GNLQSQISQT AGTIRNEITD 240
241 RTNNLQSQIT QNANNISIKV NAVGDLSNIC LNPTFIDGST EGWENVFSSS GDPGSPTKFY 300
301 GGVNTRNAFY GNMFSVAAGD KYFFSVFAWQ NQSTNPLNIG FTYLQKDGTW NWQSAINFAP 360
361 NEGARTKSGS ITIPKEAVKA RIWVEIDSFS N
|
| Detection Method: | |
| Confidence: | 10.30103 |
| Match: | 1i84S |
| Description: | Heavy meromyosin subfragment |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [1250-1333] |
1 11 21 31 41 51
| | | | | |
1 FGNWWFTNLI VRKNDTISQI NMSAGTTLIQ NDKIYMDASS TVFSGKAFIP DAAITNISAD 60
61 KINTGTLDAG RINVINLNAN NITT
|
| Detection Method: |
Shown below are all of our de novo (Rosetta) predictions for this domain.
Click here to view only most confident match.
| MCM Score |
SCOP Match |
SCOP Description | ||
| View | Download | 0.977 | b.61.3 | D-aminopeptidase, middle and C-terminal domains |
| View | Download | 0.977 | b.61.3 | D-aminopeptidase, middle and C-terminal domains |
| View | Download | 0.949 | b.61.3 | D-aminopeptidase, middle and C-terminal domains |
| View | Download | 0.949 | b.61.3 | D-aminopeptidase, middle and C-terminal domains |
| View | Download | 0.946 | b.61.8 | Description not found. |
| View | Download | 0.912 | d.82.1 | Copper amine oxidase, domain N |
| View | Download | 0.909 | b.61.8 | Description not found. |
| View | Download | 0.905 | d.82.1 | Copper amine oxidase, domain N |
| View | Download | 0.901 | d.82.1 | Copper amine oxidase, domain N |
| View | Download | 0.868 | b.1.1 | Immunoglobulin |
|
Region A: Residues: [1334-1624] |
1 11 21 31 41 51
| | | | | |
1 GTIKGQNSSF NLTDGTLTAL NQYNEGVFMR NGKLEFTSRQ SWNNNSTATY GYIQSLPKMF 60
61 ALGYNYGGLD INGTSGFVLH NDKTKIKTSN SGQSGSAVEG GSYIFGSDGL TSINNENSVI 120
121 INAGAQSYSE WTAGISLLSG NASRNVPPSI TLGSSNSLND RSTVTISGSL AVLGSKNAAV 180
181 RTSQGLRAIN AYETAEYYFG DIGRAKTNSN GAVCIYMDPL FLETVNTGVP YHIFLTSYSE 240
241 ARIWVSEMYP SYFVIRSDKP NADFVWEIKA KRKGYENDRL KIIDQEKKVN E
|
| Detection Method: | |
| Confidence: | 6.0 |
| Match: | 3cgtA |
| Description: | STRUCTURE OF CYCLODEXTRIN GLYCOSYLTRANSFERASE COMPLEXED WITH ITS MAIN PRODUCT BETA-CYCLODEXTRIN |
Matching Structure (courtesy of the PDB): |
|