| Protein: | AB11G_ARATH |
| Organism: | Arabidopsis thaliana |
| Length: | 703 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for AB11G_ARATH.
| Description | E-value | Query Range |
Subject Range |
|
|
501.0 | [0..10] | [695..2] |
|
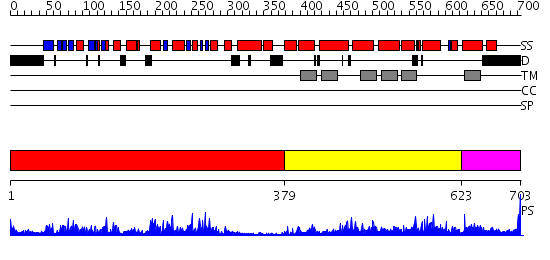
Region A: Residues: [1-378] |
1 11 21 31 41 51
| | | | | |
1 MEIEASRQQT TVPVSVGGGN FPVGGLSPLS EAIWREKAPT EFVGDVSARL TWQDLTVMVT 60
61 MGDGETQNVL EGLTGYAEPG SLTALMGPSG SGKSTMLDAL ASRLAANAFL SGTVLLNGRK 120
121 TKLSFGTAAY VTQDDNLIGT LTVRETIWYS ARVRLPDKML RSEKRALVER TIIEMGLQDC 180
181 ADTVIGNWHL RGISGGEKRR VSIALEILMR PRLLFLDEPT SGLDSASAFF VTQTLRALSR 240
241 DGRTVIASIH QPSSEVFELF DRLYLLSGGK TVYFGQASDA YEFFAQAGFP CPALRNPSDH 300
301 FLRCINSDFD KVRATLKGSM KLRFEASDDP LEKITTAEAI RLLVDYYHTS DYYYTAKAKV 360
361 EEISQFKGTI LDSGGSQA
|
| Detection Method: | |
| Confidence: | 70.154902 |
| Match: | 1z47A |
| Description: | Structure of the ATPase subunit CysA of the putative sulfate ATP-binding cassette (ABC) transporter from Alicyclobacillus acidocaldarius |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [379-622] |
1 11 21 31 41 51
| | | | | |
1 SFLLQTYTLT KRSFINMSRD FGYYWLRLLI YILVTVCIGT IYLNVGTSYS AILARGSCAS 60
61 FVFGFVTFMS IGGFPSFVED MKVFQRERLN GHYGVAAFVI ANTLSATPFL IMITFISGTI 120
121 CYFMVGLHPG FTHYLFFVLC LYASVTVVES LMMAIASIVP NFLMGIIIGA GIQGIFMLVS 180
181 GFFRLPNDIP KPFWRYPMSY ISFHFWALQG QYQNDLRGLT FDSQGSAFKI PGEYVLENVF 240
241 QIDL
|
| Detection Method: | |
| Confidence: | 29.337242 |
| Match: | PF01061.15 |
| Description: | No description for PF01061.15 was found. |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [623-703] |
1 11 21 31 41 51
| | | | | |
1 HRSKWINLSV ILSMIIIYRI IFFIMIKTNE DVTPWVRGYI ARRRMKQKNG TQNTTVAPDG 60
61 LTQSPSLRNY IATRTDGARR W
|
| Detection Method: | |
| Confidence: | 1.15 |
| Match: | 4hb1A |
| Description: | A DESIGNED FOUR HELIX BUNDLE PROTEIN. |
Matching Structure (courtesy of the PDB): |
|