| Protein: | CSLE1_ARATH |
| Organism: | Arabidopsis thaliana |
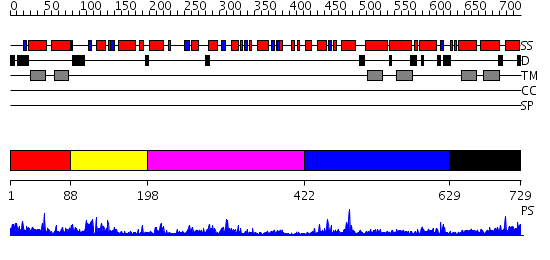
| Length: | 729 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for CSLE1_ARATH.
| Description | E-value | Query Range |
Subject Range |
|
|
595.0 | [0..6] | [729..237] |
|
Region A: Residues: [1-87] |
1 11 21 31 41 51
| | | | | |
1 MVNKDDRIRP VHEADGEPLF ETRRRTGRVI AYRFFSASVF VCICLIWFYR IGEIGDNRTV 60
61 LDRLIWFVMF IVEIWFGLYW VVTQSSR
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [88-197] |
1 11 21 31 41 51
| | | | | |
1 WNPVWRFPFS DRLSRRYGSD LPRLDVFVCT ADPVIEPPLL VVNTVLSVTA LDYPPEKLAV 60
61 YLSDDGGSEL TFYALTEAAE FAKTWVPFCK KFNVEPTSPA AYLSSKANCL
|
| Detection Method: | |
| Confidence: | 2.08 |
| Match: | 2ffuA |
| Description: | Crystal Structure of Human ppGalNAcT-2 complexed with UDP and EA2 |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [198-421] |
1 11 21 31 41 51
| | | | | |
1 DSAAEEVAKL YREMAARIET AARLGRIPEE ARVKYGDGFS QWDADATRRN HGTILQVLVD 60
61 GREGNTIAIP TLVYLSREKR PQHHHNFKAG AMNALLRVSS KITCGKIILN LDCDMYANNS 120
121 KSTRDALCIL LDEKEGKEIA FVQFPQCFDN VTRNDLYGSM MRVGIDVEFL GLDGNGGPLY 180
181 IGTGCFHRRD VICGRKYGEE EEEEESERIH ENLEPEMIKA LASC
|
| Detection Method: | |
| Confidence: | 6.69897 |
| Match: | 1h7lA |
| Description: | Spore coat polysaccharide biosynthesis protein SpsA |
Matching Structure (courtesy of the PDB): |
|
| Term | Confidence | Notes |
| catalytic activity | 1.3567084433464 | bayes_pls_golite062009 |
| transferase activity | 1.04375515050786 | bayes_pls_golite062009 |
|
Region A: Residues: [422-628] |
1 11 21 31 41 51
| | | | | |
1 TYEENTQWGK EMGVKYGCPV EDVITGLTIQ CRGWKSAYLN PEKQAFLGVA PTNLHQMLVQ 60
61 QRRWSEGDFQ IMLSKYSPVW YGKGKISLGL ILGYCCYCLW APSSLPVLIY SVLTSLCLFK 120
121 GIPLFPKVSS SWFIPFGYVT VAATAYSLAE FLWCGGTFRG WWNEQRMWLY RRTSSFLFGF 180
181 MDTIKKLLGV SESAFVITAK VAEEEAA
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [629-729] |
1 11 21 31 41 51
| | | | | |
1 ERYKEEVMEF GVESPMFLVL GTLGMLNLFC FAAAVARLVS GDGGDLKTMG MQFVITGVLV 60
61 VINWPLYKGM LLRQDKGKMP MSVTVKSVVL ALSACTCLAF L
|
| Detection Method: |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.