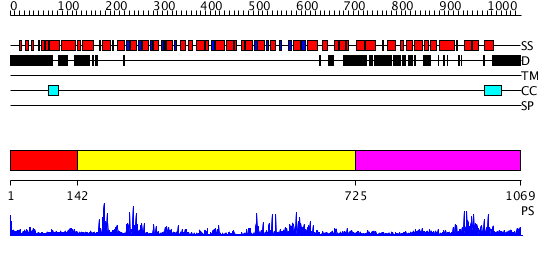
| Protein: | CHR17_ARATH |
| Organism: | Arabidopsis thaliana |
| Length: | 1069 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
No multiple sequence alignment data found for CHR17_ARATH.
|
Region A: Residues: [1-141] |
1 11 21 31 41 51
| | | | | |
1 MARASKREVS SDEAYSSEEE EQVNDQANVE EDDDELEAVA RSAGSDEEDV APDEAPVSDD 60
61 EVVPVEDDAE EDEEDEEKAE ISKREKARLK EMQKMKKQKI QQILDSQNAS IDADMNNKGK 120
121 GRIKYLLQQT ELFAHFAKSD P
|
| Detection Method: | |
| Confidence: | 13.154902 |
| Match: | 2b2uA |
| Description: | Tandem chromodomains of human CHD1 complexed with Histone H3 Tail containing trimethyllysine 4 and dimethylarginine 2 |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [142-724] |
1 11 21 31 41 51
| | | | | |
1 SPSQKKGKGR GRHSSKLTEE EEDEECLKEE EGGIVGSGGT RLLTQPACIQ GKLRDYQLAG 60
61 LNWLIRLYEN GINGILADEM GLGKTLQTIS LLAYLHEYRG INGPHMVVAP KSTLGNWMNE 120
121 IRRFCPVLRA VKFLGNPEER RHIREELLVA GKFDICVTSF EMAIKEKTTL RRFSWRYIII 180
181 DEAHRIKNEN SLLSKTMRLF STNYRLLITG TPLQNNLHEL WALLNFLLPE VFSSAETFDE 240
241 WFQISGENDQ QEVVQQLHKV LRPFLLRRLK SDVEKGLPPK KETILKVGMS QMQKQYYKAL 300
301 LQKDLEVVNG GGERKRLLNI AMQLRKCCNH PYLFQGAEPG PPYTTGDHLV TNAGKMVLLD 360
361 KLLPKLKDRD SRVLIFSQMT RLLDILEDYL MYRGYQYCRI DGNTGGDERD ASIEAYNKPG 420
421 SEKFVFLLST RAGGLGINLA TADVVILYDS DWNPQVDLQA QDRAHRIGQK KEVQVFRFCT 480
481 ENAIEAKVIE RAYKKLALDA LVIQQGRLAE QKTVNKDELL QMVRYGAEMV FSSKDSTITD 540
541 EDIDRIIAKG EEATAELDAK MKKFTEDAIQ FKMDDSADFY DFD
|
| Detection Method: | |
| Confidence: | 89.69897 |
| Match: | 1z3iX |
| Description: | Structure of the SWI2/SNF2 chromatin remodeling domain of eukaryotic Rad54 |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [725-1069] |
1 11 21 31 41 51
| | | | | |
1 DDNKDESKVD FKKIVSENWN DPPKRERKRN YSEVEYFKQT LRQGAPAKPK EPRIPRMPQL 60
61 HDFQFFNIQR LTELYEKEVR YLMQAHQKTQ MKDTIEVDEP EEVGDPLTAE EVEEKELLLE 120
121 EGFSTWSRRD FNAFIRACEK YGRNDIKSIA SEMEGKTEEE VERYAQVFQV RYKELNDYDR 180
181 IIKNIERGEA RISRKDEIMK AIGKKLDRYR NPWLELKIQY GQNKGKLYNE ECDRFMICMV 240
241 HKLGYGNWDE LKAAFRTSPL FRFDWFVKSR TTQELARRCD TLIRLIEKEN QEFDERERQA 300
301 RKEKKLSKSA TPSKRPSGRQ ANESPSSLLK KRKQLSMDDY GKRRK
|
| Detection Method: | |
| Confidence: | 43.39794 |
| Match: | 1ofcX |
| Description: | nucleosome recognition module of ISWI ATPase |
Matching Structure (courtesy of the PDB): |
|