| Protein: | HSFA2_ARATH |
| Organism: | Arabidopsis thaliana |
| Length: | 345 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for HSFA2_ARATH.
| Description | E-value | Query Range |
Subject Range |
|
|
241.0 | [0..15] | [315..1] |
|
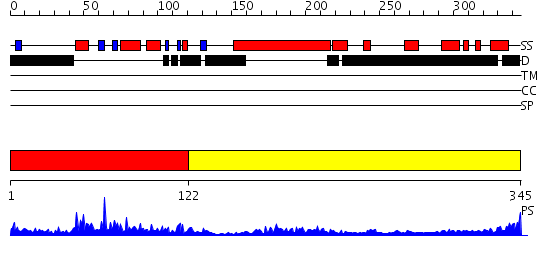
Region A: Residues: [1-121] |
1 11 21 31 41 51
| | | | | |
1 MEELKVEMEE ETVTFTGSVA ASSSVGSSSS PRPMEGLNET GPPPFLTKTY EMVEDPATDT 60
61 VVSWSNGRNS FVVWDSHKFS TTLLPRYFKH SNFSSFIRQL NTYGFRKIDP DRWEFANEGF 120
121 L
|
| Detection Method: | |
| Confidence: | 29.39794 |
| Match: | 1hksA |
| Description: | SOLUTION STRUCTURE OF THE DNA-BINDING DOMAIN OF DROSOPHILA HEAT SHOCK TRANSCRIPTION FACTOR |
Matching Structure (courtesy of the PDB): |
|
| Term | Confidence | Notes |
| binding | 0.835523092977445 | bayes_pls_golite062009 |
| transcription regulator activity | 0.4843036104266 | bayes_pls_golite062009 |
| nucleic acid binding | 0.37117756729668 | bayes_pls_golite062009 |
| DNA binding | 0.3118855114645 | bayes_pls_golite062009 |
|
Region A: Residues: [122-345] |
1 11 21 31 41 51
| | | | | |
1 AGQKHLLKNI KRRRNMGLQN VNQQGSGMSC VEVGQYGFDG EVERLKRDHG VLVAEVVRLR 60
61 QQQHSSKSQV AAMEQRLLVT EKRQQQMMTF LAKALNNPNF VQQFAVMSKE KKSLFGLDVG 120
121 RKRRLTSTPS LGTMEENLLH DQEFDRMKDD MEMLFAAAID DEANNSMPTK EEQCLEAMNV 180
181 MMRDGNLEAA LDVKVEDLVG SPLDWDSQDL HDMVDQMGFL GSEP
|
| Detection Method: | |
| Confidence: | 4.39794 |
| Match: | 1i84S |
| Description: | Heavy meromyosin subfragment |
Matching Structure (courtesy of the PDB): |
|