| Protein: | gi|110743660, gi... |
| Organism: | Arabidopsis thaliana |
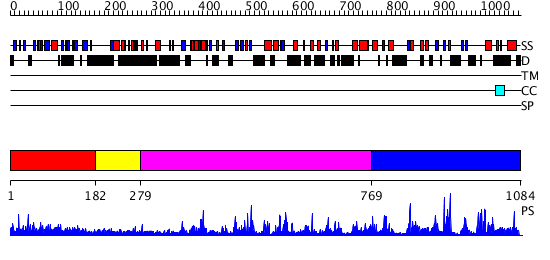
| Length: | 1084 amino acids |
| Reference: | Drew K, et al. (2011) The proteome folding project: Proteome-scale prediction of structure and function. Genome Res. 2011 Sep 16 |
Listed below are up to the top 10 sequence alignment matches, by species, for the PSI-BLAST search against the protein sequence for gi|110743660, gi....
| Description | E-value | Query Range |
Subject Range |
|
|
1077.0 | [0..1] | [1076..1] |
|
|
843.0 | [0..9] | [1081..23] |
|
|
655.0 | [0..114] | [1079..92] |
|
|
435.0 | [0..233] | [761..29] |
|
|
429.0 | [0..232] | [761..24] |
|
Region A: Residues: [1-181] |
1 11 21 31 41 51
| | | | | |
1 MKRRQCEKVV IRIHNIGTPL ISGSSGLPLE LFHIQSDRPY TIGRSSSDGF CDFVIDHSSI 60
61 SRKHCQILFD SQSHKLYIFD GVIHLPSGSF SQVYDEFRRR LVGVEDLGNL KFRASLNGVY 120
121 VNRVRVRKSK VQEVSIDDEV LFFCGKEGLC CKDGRVGFVV QEIVFEGRDA SIVSVSSGHS 180
181 R
|
| Detection Method: | |
| Confidence: | 13.79588 |
| Match: | PF00498.18 |
| Description: | No description for PF00498.18 was found. |
Shown below is our most confident prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [182-278] |
1 11 21 31 41 51
| | | | | |
1 GTFSSGKRSK RVFAPMENEI NSPVSGFYPP KAVGVVERVN SLVSYCRHIL KSDDPLSCLR 60
61 LSIISHSGKE CLSCCTSKMF RSKVGIVADD RGVKSAE
|
| Detection Method: |
Shown below is our most confident de novo (Rosetta) prediction for this domain.
Click here to view all matches.
Found no confident structure predictions for this domain.
|
Region A: Residues: [279-768] |
1 11 21 31 41 51
| | | | | |
1 INHDMGHGLS GLRLSIERPN SNLHVDRRLG VSDLISEIEN EFAACTFISD KTRTMLPFDG 60
61 EKVNTPDITC INKEKSYQSS LQAPGKNFYL NRLQYIEQSS TGCQRVVSLP ELLHPVESIQ 120
121 QIFLATFTSD ILWFLTCCDT PRHLPVTIAC HNAERCWSSN PDARTAVPLP NYPNVTMVYP 180
181 PFPEEIAFGK DRTNRGIACH HPKLFILQRK DSIRVIITSA NLVARQWNDV TNTVWWQDFP 240
241 RRADPDLLSL FGHCQRETNH GLKPDFCAQL AGFAASLLTD VPSQAHWILE FTKYNFEHSA 300
301 GHLVASVPGI HSYKPSYLTE SGCSNTIFSE EFLGSVEASV VGLSYLFRSA NDSTGAQLKR 360
361 LASYIRRTRE NSLGMLELVL RRNTNVPADP NAVRVLVPNP DDDSRDDFVQ LGFLPRSIAK 420
421 WVSPLWDIGF FKFVGYVYRD EVLGAASCRS NEKVQLVLHV LQGVSISDMS KLIQPYHVVA 480
481 LCSLIASLQR
|
| Detection Method: | |
| Confidence: | 50.69897 |
| Match: | 1q32A |
| Description: | Crystal Structure Analysis of the Yeast Tyrosyl-DNA Phosphodiesterase |
Matching Structure (courtesy of the PDB): |
|
|
Region A: Residues: [769-1084] |
1 11 21 31 41 51
| | | | | |
1 CTGIWRLQEV LGRYKWPESQ ESDFVYSASS IGGSATTGFQ ADFSSAAGKK ALQHFDSQES 60
61 DPEWGCWSNR EEREAPSIKI IFPTIERVKN GHHGVLSSRR LLCFSEKTWQ KWRHSNVLHD 120
121 AVPNPQDRVG HPMHIKVARR LFTSTRGSRS SSFGWVYSGS HNFSAAAWGQ TISRSSRNNQ 180
181 DQSNNAIRAV KKLRVCNYEL GIVFVFPPPH EETDSCEGSK IDDIVLPFVV PAPKYGWSDK 240
241 PATGLAMREA FAEFREGSTS FCGESEVEEE VEEEEEEEAD AEGRGEFVAE EEKQEEEAYA 300
301 EALWSQVESS SSSLSS
|
| Detection Method: | |
| Confidence: | 102.0 |
| Match: | 1qzqA |
| Description: | human Tyrosyl DNA phosphodiesterase |
Matching Structure (courtesy of the PDB): |
|